


There are only currently two main drugs used to attempt to treat ALS and prolong the life of the diagnosed individual. Riluzole is a glutamate release inhibitor that has been FDA approved for over twenty years.5 Riluzole can also interfere with the creation of reactive oxygen species, such as superoxide anions. Alongside the ability of Riluzole to inhibit the release of glutamate, it also enhances the ability to reuptake glutamate from the synapse. One study has found Riluzole to extend the average life span of an individual diagnosed with ALS from slightly under twelve months to slightly under fifteen months.6 This slight increase in average lifespan resulted in a nine percent increase in individuals living a year past diagnosis.
The second drug used for the treatment of ALS is Edaravone. Edaravone is used to help counteract the adverse effects of free radicals to reduce oxidative stress on neurological cells.7 To evaluate the effectiveness of the treatment using Edaravone, the Revised Amyotrophic Lateral Sclerosis Functioning Rate Scale was used (ALSFRS-R). This scale offers a variety of tests to observe the decline in function of people suffering from ALS. Edaravone has been shown to significantly improve the ALSFRS-R scores at six and twelve months after diagnosis of ALS.8 This improvement of ALSFRS-R scores represents the improvement of the quality of life of individuals diagnosed with ALS.
Although these drugs have shown an increase in the span and quality of life for patients with ALS, there is no known cure or treatment that allows for a long-term or potentially complete cure. From this predicament of the unknowns behind the causes and advancement of ALS, there are multiple theories hypothesizing the mechanisms of ALS. Due to advancements in biotechnology, comparative analysis of genes has allowed for a better understanding of the potential causes of ALS. The most common mutated genes observed within individuals suffering from ALS are SOD1, C9ORF72, FUS, and TARDBP; however, this can vary depending on the continent being examined within the study.9 As seen in the image below, the believed causes of fALS and sALS differ drastically from one another.10 It is also important to note that the fALS causative mutations differ greatly based on the population that is being assessed. This could be a result of a mutated gene being represented at a higher probability in different populations across the globe.
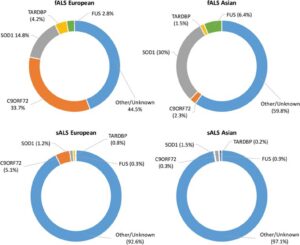
Superoxide dismutase one (SOD1) is a commonly mutated enzyme in cases of fALS.10 This enzyme is responsible for the rapid disproportionation of superoxide (O2-) into hydrogen peroxide (H2O2).14 This enzyme is a homodimer consisting of two identical subunits.15 Each identical subunit consists of an eight stranded Greek key beta-barrel fold. These barrels contain binding sites for one zinc ion and one copper ion. The copper ion is used throughout the disproportionation reaction to convert the superoxide into hydrogen peroxide. The zinc ion is believed to be important for the overall stability of the subunit.
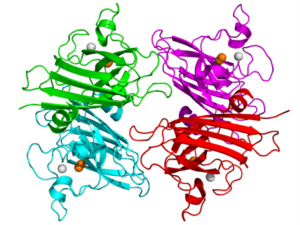
Two theories have arisen to be the hypothesized causes of this form of SOD1 affiliated fALS. Initially, studies of SOD1 affiliated fALS showed a loss of function of over fifty percent of the SOD1 enzymatic activity based on the relative mutation.17 This lack of enzymatic activity was believed to cause a buildup of superoxide within the cell. Superoxide, which is a normal byproduct formed during cellular respiration, was believed to cause oxidative damage to the neuron resulting in the mechanism causing the slow neurodegeneration of the individual.14 The second and more proposed theory behind SOD1 fALS is the toxic gain of function of the mutated SOD1.19 This toxic gain of function has shown that it could result in the motor neuron neurodegenerative disease without alteration of the protein SOD1 or its activity levels.
Mutation within the TARDBP (TAR DNA-binding protein) gene has also been shown to have an affiliation and potential cause of ALS.20 The TARDBP is responsible for encoding the TAR DNA-binding protein 43 (TDP-43).21 This protein is comprised of a four hundred fourteen amino acid sequence.22 This sequence contains a nuclear localization signal and nuclear export signal allowing to shuttle in and out of the nucleus. The purpose of this protein is to influence gene expression through manipulation of mRNA and pre-mRNA.23
Particularly with cases of sALS, it is hypothesized that there are multiple factors rather than a single event responsible for the cause and aggression of the case.10 The amount of causes that have been discovered leading to sALS represent that there are many avenues on the molecular level that can result in neurodegeneration. Ten of the most common disease causing mechanisms can be observed below.
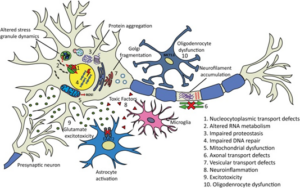
Studies of all causes of ALS are essential to solving each individual case. Having the ability to cure SOD1 or TDP-43 affiliated fALS could lead to a breakthrough in the entire field of ALS research. ALS is the most prevalent of all motor neuron diseases with an incidence rate of roughly two out of every one hundred thousand people.3 The prevalence alongside the unknown cause of this disease goes to represent that neurological disorders are very difficult in determining the cause. Currently, the development of RNA therapeutics with short interfering RNA(siRNA) and antisense oligonucleotides (AO) are believed to be the most promising route of potential cures to ALS causes.
There is much to learn about fALS, sALS, and ALS. Research is drastically needed to uncover the connection and commonalities between the unknown and common causes. The drugs and treatments being offered provide some sort of insight into potential methods of treatment; however, as seen stated above, these drugs only give slight delay to the inevitable end of the fight against ALS. Based on recent advancements in genetic sequencing, analysis, and RNA therapeutics, one can only hope that a breakthrough can be found associated with ALS and other motor neuron degenerative diseases.
- “Babe Ruth & Lou Gehrig.” Openverse, https://openverse.org/image/cad0c5af-b7a2-459d-b38f-ad13e5c18ac9?q=lou+gehrig. ↵
- Fred H. L. (2017). Lou Gehrig and Ed Todd: Greatness Interrupted. Texas Heart Institute journal, 44(6), 383–384. https://doi.org/10.14503/THIJ-17-6540. ↵
- Chiò, A., Logroscino, G., Traynor, B. J., Collins, J., Simeone, J. C., Goldstein, L. A., & White, L. A. (2013). Global epidemiology of amyotrophic lateral sclerosis: a systematic review of the published literature. Neuroepidemiology, 41(2), 118–130. https://doi.org/10.1159/000351153. ↵
- Alsultan, A. A., Waller, R., Heath, P. R., & Kirby, J. (2016). The genetics of amyotrophic lateral sclerosis: current insights. Degenerative neurological and neuromuscular disease, 6, 49–64. https://doi.org/10.2147/DNND.S84956. ↵
- Blyufer, A., Lhamo, S., Tam, C., Tariq, I., Thavornwatanayong, T., & Mahajan, S. S. (2021). Riluzole: A neuroprotective drug with potential as a novel anti cancer agent (Review). International journal of oncology, 59(5), 95. https://doi.org/10.3892/ijo.2021.5275. ↵
- Miller, R. G., Mitchell, J. D., & Moore, D. H. (2012). Riluzole for amyotrophic lateral sclerosis (ALS)/motor neuron disease (MND). The Cochrane database of systematic reviews, 2012(3), CD001447. https://doi.org/10.1002/14651858.CD001447.pub3. ↵
- Park, J. M., Kim, S. Y., Park, D., & Park, J. S. (2020). Effect of edaravone therapy in Korean amyotrophic lateral sclerosis (ALS) patients. Neurological sciences : official journal of the Italian Neurological Society and of the Italian Society of Clinical Neurophysiology, 41(1), 119–123. https://doi.org/10.1007/s10072-019-04055-3. ↵
- Okada, M., Yamashita, S., Ueyama, H., Ishizaki, M., Maeda, Y., & Ando, Y. (2018). Long-term effects of edaravone on survival of patients with amyotrophic lateral sclerosis. eNeurologicalSci, 11, 11–14. https://doi.org/10.1016/j.ensci.2018.05.001. ↵
- Boylan K. (2015). Familial Amyotrophic Lateral Sclerosis. Neurologic clinics, 33(4), 807–830. https://doi.org/10.1016/j.ncl.2015.07.001. ↵
- Mejzini, R., Flynn, L. L., Pitout, I. L., Fletcher, S., Wilton, S. D., & Akkari, P. A. (2019). ALS Genetics, Mechanisms, and Therapeutics: Where Are We Now?. Frontiers in neuroscience, 13, 1310. https://doi.org/10.3389/fnins.2019.01310. ↵
- Mejzini, R., Flynn, L. L., Pitout, I. L., Fletcher, S., Wilton, S. D., & Akkari, P. A. (2019). ALS Genetics, Mechanisms, and Therapeutics: Where Are We Now?. Frontiers in neuroscience, 13, 1310. https://doi.org/10.3389/fnins.2019.01310. ↵
- Mejzini, R., Flynn, L. L., Pitout, I. L., Fletcher, S., Wilton, S. D., & Akkari, P. A. (2019). ALS Genetics, Mechanisms, and Therapeutics: Where Are We Now?. Frontiers in neuroscience, 13, 1310. https://doi.org/10.3389/fnins.2019.01310. ↵
- Mejzini, R., Flynn, L. L., Pitout, I. L., Fletcher, S., Wilton, S. D., & Akkari, P. A. (2019). ALS Genetics, Mechanisms, and Therapeutics: Where Are We Now?. Frontiers in neuroscience, 13, 1310. https://doi.org/10.3389/fnins.2019.01310. ↵
- Tainer, J., Getzoff, E., Richardson, J. et al. Structure and mechanism of copper, zinc superoxide dismutase. Nature 306, 284–287 (1983). https://doi.org/10.1038/306284a0. ↵
- Clara, M., N., C., H., F., & G., A. (2013). Superoxide Dismutase and Oxidative Stress in Amyotrophic Lateral Sclerosis. InTech. doi: 10.5772/56488. ↵
- “Copper, Zinc Superoxide Dismutase.” Openverse, https://openverse.org/image/5719a80b-c266-45d9-b588-2b4e9125508c?q=superoxide+dismutase. ↵
- Deng HX, Hentati A, Tainer JA, Iqbal Z, Cayabyab A, Hung WY, Getzoff ED, Hu P, Herzfeldt B, Roos RP, et al. Amyotrophic lateral sclerosis and structural defects in Cu,Zn superoxide dismutase. Science. 1993 Aug 20;261(5124):1047-51. doi: 10.1126/science.8351519. PMID: 8351519. ↵
- Tainer, J., Getzoff, E., Richardson, J. et al. Structure and mechanism of copper, zinc superoxide dismutase. Nature 306, 284–287 (1983). https://doi.org/10.1038/306284a0. ↵
- Bruijn, L.I. M.W. Becher, M.K. Lee, K.L. Anderson, N.A. Jenkins, N.G. Copeland, S.S. Sisodia, J.D. Rothstein, D.R. Borchelt, D.L. Price, D.W. Cleveland, (1997). ALS-Linked SOD1 Mutant G85R Mediates Damage to Astrocytes and Promotes Rapidly Progressive Disease with SOD1-Containing Inclusions, Neuron, Volume 18, Issue 2, 1997, Pages 327-338, ISSN 0896-6273, https://doi.org/10.1016/S0896-6273(00)80272-X. ↵
- Leigh PN, Whitwell H, Garofalo O, Buller J, Swash M, Martin JE, Gallo JM, Weller RO, Anderton BH. Ubiquitin-immunoreactive intraneuronal inclusions in amyotrophic lateral sclerosis. Morphology, distribution, and specificity. Brain. 1991 Apr;114 ( Pt 2):775-88. doi: 10.1093/brain/114.2.775. PMID: 1646064. ↵
- Arai T, Hasegawa M, Akiyama H, Ikeda K, Nonaka T, Mori H, Mann D, Tsuchiya K, Yoshida M, Hashizume Y, Oda T. TDP-43 is a component of ubiquitin-positive tau-negative inclusions in frontotemporal lobar degeneration and amyotrophic lateral sclerosis. Biochem Biophys Res Commun. 2006 Dec 22;351(3):602-11. doi: 10.1016/j.bbrc.2006.10.093. Epub 2006 Oct 30. PMID: 17084815. ↵
- Ayala YM, Zago P, D’Ambrogio A, Xu YF, Petrucelli L, Buratti E, Baralle FE. Structural determinants of the cellular localization and shuttling of TDP-43. J Cell Sci. 2008 Nov 15;121(Pt 22):3778-85. doi: 10.1242/jcs.038950. Epub 2008 Oct 28. PMID: 18957508. ↵
- Buratti E, Baralle FE. The multiple roles of TDP-43 in pre-mRNA processing and gene expression regulation. RNA Biol. 2010 Jul-Aug;7(4):420-9. doi: 10.4161/rna.7.4.12205. Epub 2010 Jul 1. PMID: 20639693. ↵
- Mejzini, R., Flynn, L. L., Pitout, I. L., Fletcher, S., Wilton, S. D., & Akkari, P. A. (2019). ALS Genetics, Mechanisms, and Therapeutics: Where Are We Now?. Frontiers in neuroscience, 13, 1310. https://doi.org/10.3389/fnins.2019.01310. ↵
- Mejzini, R., Flynn, L. L., Pitout, I. L., Fletcher, S., Wilton, S. D., & Akkari, P. A. (2019). ALS Genetics, Mechanisms, and Therapeutics: Where Are We Now?. Frontiers in neuroscience, 13, 1310. https://doi.org/10.3389/fnins.2019.01310. ↵
- Chiò, A., Logroscino, G., Traynor, B. J., Collins, J., Simeone, J. C., Goldstein, L. A., & White, L. A. (2013). Global epidemiology of amyotrophic lateral sclerosis: a systematic review of the published literature. Neuroepidemiology, 41(2), 118–130. https://doi.org/10.1159/000351153. ↵


1 comment
Osondra Fournier-Colon
Congratulations on your nomination; this article is such a great awareness spreader to ALS and the treatment there are for it. This article does a great dive into the history of an illness and gives the reader a good background on the matter. I like how you describe the biology behind the illness and how it works within your body. It is comforting to know how an illness works.