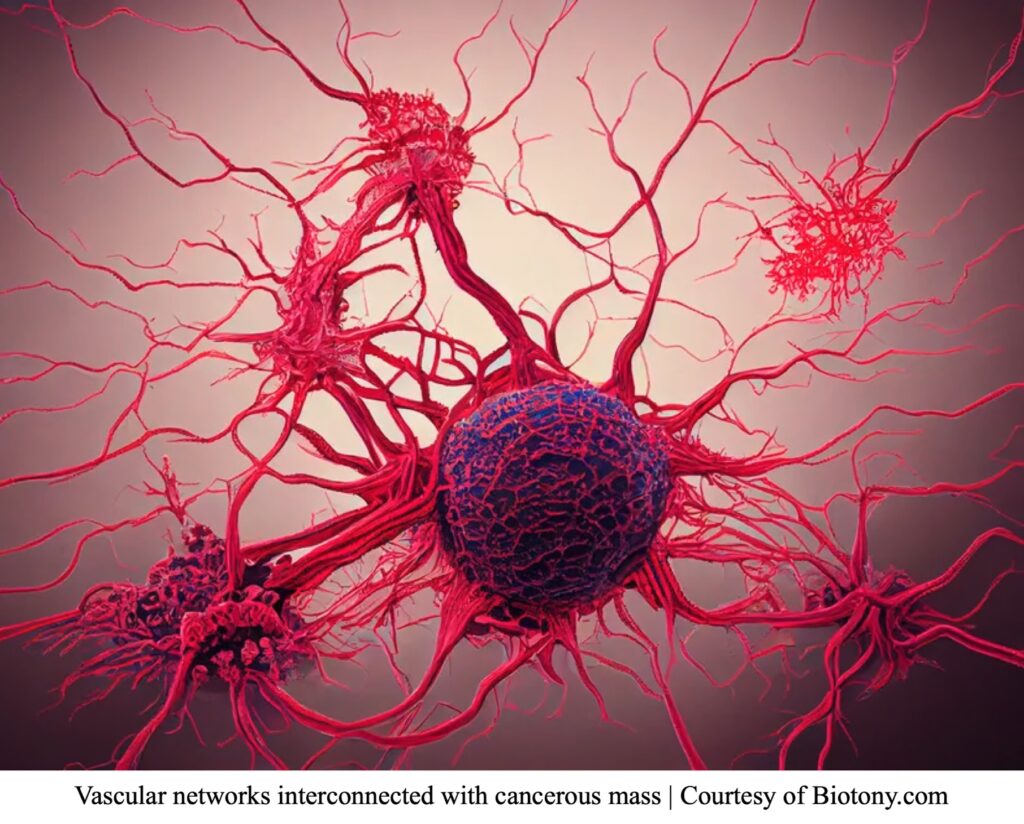

Cancer may be one of the most feared diseases. Many have been affected by cancer, whether through a loved one or maybe even yourself. Often, we ask the question, “Why us?” or “Why them?” in search of a higher, more spiritual answer. When we follow the journey of a loved one facing a disease as overpowering as cancer, we see that their health quickly deteriorates due to the ongoing physical symptoms, even with treatment, but why is that?
The National Cancer Institute defines cancer as “…a disease in which some of the body’s cells grow uncontrollably and spread to other parts of the body…”.1 The emergence of cancer is influenced by numerous pathways. Sometimes, it is due to carcinogen exposure, rising issues in DNA replication, or a combination of both. To understand how cancer progresses, it is necessary to understand how a cell can continue to proliferate and survive, as well as how it increases its nutrients to support the growing tumor mass. One prime example of abnormal angiogenesis is Kaposi’s Sarcoma, but that will be discussed later.
When we mention DNA replication and its biomechanics, what exactly does that entail? DNA replication is a natural process in which our cells undergo cellular division. This takes roughly an hour to complete.2 This process is essential for our well-being because it allows our body to grow tissue and heal from previous wounds or diseases. When a cell divides, it will duplicate chromosomes, proteins, nucleotides, organelles, and its size to produce two daughter cells.3 This process is called the cell cycle, and it has four main stages to it. The most important cell cycle process for this article is interphase, which will be where the following phases will take place. The first cell growth stage is the G1 phase, where the cell will duplicate in size by stimulating protein and nucleotide synthesis. Everything duplicated in G1 will be mainly used in S-phase, where the nucleus is replicated. The G2 phase is where DNA replication occurs, meaning that the whole genome of a cell is copied to make duplicates.4 Nucleotides duplicated during G1 will now be used to build the new genome for the daughter cells. Once DNA replication has been completed, the cell proceeds to the G2 phase.
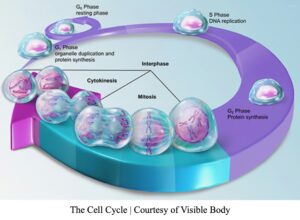 Knowing all this, you probably ask yourself, are there any checkpoints during cell division to prevent damaged DNA from being passed down to new daughter cells? Several proteins play a security role in this cellular process. The tumor protein p53 is known to be a gatekeeper tumor suppressor gene that halts cell division in G1 if there are any issues in cell size, lack of nutrient and growth factor availability, and lastly, DNA damage. If such an issue is seen in cells undergoing the G1 phase, then p53 halts cell division by sending the damaged cells to the G0 phase. This phase stops cell division until the cells can fix the issues encountered by p53. The p53 protein will activate DNA damage repair proteins to fix cells that have DNA damage due to base dimerization or incorrect or missing nucleotide base pairing. If cells fail to resolve issues encountered, p53 triggers programmed cell death, also known as apoptosis. The overall role of p53 is to trigger cell arrest or apoptosis to halt cell proliferation, therefore serving as the “guardian of the genome”.5
Knowing all this, you probably ask yourself, are there any checkpoints during cell division to prevent damaged DNA from being passed down to new daughter cells? Several proteins play a security role in this cellular process. The tumor protein p53 is known to be a gatekeeper tumor suppressor gene that halts cell division in G1 if there are any issues in cell size, lack of nutrient and growth factor availability, and lastly, DNA damage. If such an issue is seen in cells undergoing the G1 phase, then p53 halts cell division by sending the damaged cells to the G0 phase. This phase stops cell division until the cells can fix the issues encountered by p53. The p53 protein will activate DNA damage repair proteins to fix cells that have DNA damage due to base dimerization or incorrect or missing nucleotide base pairing. If cells fail to resolve issues encountered, p53 triggers programmed cell death, also known as apoptosis. The overall role of p53 is to trigger cell arrest or apoptosis to halt cell proliferation, therefore serving as the “guardian of the genome”.5
During the second growth phase, the G2 phase, the cell is evaluated for p53 to ensure that cell size is correct and DNA replication is fully completed. Additionally, the DNA’s integrity is assessed. These checkpoints are in place to limit the inheritance of damaged DNA to future cells. If daughter cells inherit a damaged genome, an individual is more susceptible to disease. The result would be an increase in mutations such as inversions, duplications, frameshifts, and translocations.
Interphase is a crucial step in the cell cycle because it will dictate the course of cell division. When there are issues presented in DNA replication and p53 cannot detect them, the cell proliferation rate becomes excessive. Cells begin to divide at an increased rate and take up more space than normal cells. Uncontrolled cell division leads to the development of a tumor. However, not all tumor masses cause injury to the body. The first is a benign tumor, a non-cancerous mass that does not invade neighboring cells or metastasize. With time, it may become malignant. Sometimes, proto-oncogenes, which are genes that induce cell division but are non-cancerous, influence the growth of benign tumors. On the contrary, a malignant tumor is caused by oncogenes promoting uncontrolled cell proliferation. Malignant tumors are invasive and can travel to other parts of the body through the circulatory and lymphatic systems. These tumors often arise when issues are presented in the G1 and G2 phases due to mutations in the tumor suppressor gene, p53. The p53 protein has several functions: it regulates tumor growth by placing the brakes on cellular proliferation. However, when DNA damage or mechanical issues arise in cell division, the cells begin to divide at an increased rate, eventually leading to a mass. That is a result of p53 not being able to fulfill its job of detecting damaged DNA and activating repair mechanisms.
We mentioned that the circulatory system is involved in tumor migration, but how? For starters, the circulatory system is a complex network of blood vessels with many channels. These channels are referred to as blood vessels. There are three types of blood vessels: arteries, veins, and capillaries. Arteries carry oxygenated blood from the heart to the rest of the body. In contrast, veins carry deoxygenated blood from the body to the heart. Lastly, capillaries connect arteries and veins to fulfill this transaction.6 Blood vessel channels are essential for distributing oxygen and nutrients to tissues throughout the body. Since nutrients are carried in the blood, tumors can induce angiogenesis, a process involving the formation of new blood vessels by sprouting from previously established vessels.7
During embryo development, the embryonic stem cells begin to differentiate into endothelial cells, forming the lining of blood vessels. Vasculogenesis is the process of organizing endothelial cells into “…a primitive network of channels…”.8 The organization of the endothelial cells will eventually allow channels to be used for blood distribution. Then, angiogenesis induces the growth of bud-like structures. These bud structures will prop outwards from the main vessel, eventually leading to smaller vessels. Its branched-out motion allows development to occur in any area of the vessels when triggering growth factors are released. The exact mechanism properties of cancer-stimulating angiogenesis are still unknown. However, several studies have concluded that cancer cells release growth factors (Vascular Endothelial Growth Factor (VEGF), Fibroblast Growth factors (FGF), or others), which can diffuse through tiny pores of nearby vessels. Matrix Metalloproteinases (MMP) proteins degrade the extracellular matrix (ECM) and basal lamina components when these molecules are released. Degrading the basement support of normal cells allows for endothelial cells of the vessels to become active and undergo angiogenesis. This will eventually lead to the growth and elongation of the blood vessels. Once cells have divided to elongate and create more channels, they can begin to grow toward the tumor. Note that angiogenesis is a highly regulated biomechanical process and is thereby only activated at certain stages of life. 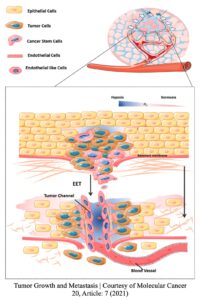 When a tumor does not have access to blood supply, it can only grow between 1-2 millimeters (mm) in size. That is the size of a sharpened pencil tip! Also, the small tumor breaks into tiny particles to release in the bloodstream to find a new growing location. It does so since its growth is “maxed out” due to a lack of supply.9 Conversely, a tumor can grow larger than 2 mm if it has access to a blood supply. For a tumor with blood supply, there is no size limit. With generations of research, the science and medical community have concluded that restricting the blood supply will limit cancer progression and its invasion attributes.
When a tumor does not have access to blood supply, it can only grow between 1-2 millimeters (mm) in size. That is the size of a sharpened pencil tip! Also, the small tumor breaks into tiny particles to release in the bloodstream to find a new growing location. It does so since its growth is “maxed out” due to a lack of supply.9 Conversely, a tumor can grow larger than 2 mm if it has access to a blood supply. For a tumor with blood supply, there is no size limit. With generations of research, the science and medical community have concluded that restricting the blood supply will limit cancer progression and its invasion attributes.
Cancerous cells can also promote angiogenesis and become invasive. When a cancer becomes invasive, it directly migrates and penetrates neighboring tissues. Meaning it infects nearby cells with its oncogenic properties and makes them cancerous. Often, with tumor invasion, there is metastasis, which is the ability of a cancer cell to enter the bloodstream and find another location to form new tumors. Nonetheless, a direct blood supply is necessary for a malignant tumor to experience enhanced growth, invasion, and metastasis.10 The blood’s nutrients make cancer strong, allowing such pathways to be activated. MMP proteins play a role in metastasis, facilitating the basal lamina and ECM destruction.11 When a cancerous tumor begins to metastasize, it penetrates through the walls of the blood vessels to gain access to the bloodstream. As the heart starts to pump blood through the arteries and the veins carry the deoxygenated blood back to the heart, the cancerous cells move through the circulatory system. Often, the cancerous cells get stuck at capillaries because their pores are only the size of a normal cell. Cancer cells have a bigger circumference than normal cells due to their oncogenic properties, forcing them to be trapped. If a cancer cell is stuck at a capillary, it begins metastases, which involves the formation of a secondary cancer mass. The secondary cancer may grow larger than the primary tumor while also developing into a different kind of cancer. It is essential to know that secondary tumors are not always successful during metastasis. Not all cancer cells survive in the bloodstream environment; only a few cells can adapt to the atmosphere and find new locations for metastasizing.
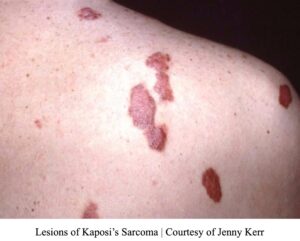
Knowing everything we have covered, how can we apply these concepts to a disease such as Kaposi’s Sarcoma? Kaposi’s Sarcoma (KS) is a disease derived from HIV/AIDS and is often only seen in those infected with the KSHV (Kaposi’s sarcoma-associated herpesvirus).12 As is well known, individuals with the Human Immunodeficiency Virus (HIV) are often predisposed to developing symptoms of Acquired Immunodeficiency Syndrome (AIDS) due to the weakening of the immune system. A few diseases that affect HIV-only individuals are anal cancers, tuberculosis, isosporiasis (parasite-induced illness), and pneumonia.13 Individuals are at a greater risk of developing KS because it is one of the more aggressive diseases that can affect individuals with HIV/AIDS. Other diseases entail the development of lymphomas and cervical cancers.14
Kaposi’s sarcoma is the tumor of blood vessels; therefore, it is abnormal angiogenesis. The blood vessels have become cancerous by invading, metastasizing, and being aggressive towards the body. It causes overproduction of blood vessels, therefore leading to red, brown, or purple skin lesions.15 The color and number of lesions depend on the aggressiveness of the cancer.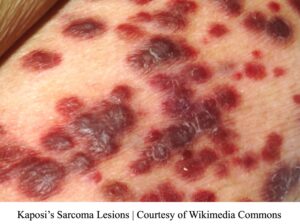
KS is caused by KSHV, a sexually transmitted virus. It is also termed human herpesvirus-8 (HHV-8).16, 17 The KSHV virus directly targets specific cells to stimulate tumor development. KSHV alone is insufficient to cause cancer; it needs a deliberate immune system. Hence, KSHV must partner with AIDS. Patients with KS may also develop lymphomas and cervical cancers induced by the Human Papilloma Virus (HPV). Lymphomas are often caused by the Epstein-Bar virus (EBV) or KSHS. Liver and cervical cancers may be triggered by Hepatitis-B (HBV) or Hepatitis-C (HCV) as well as environmental factors.18, 19
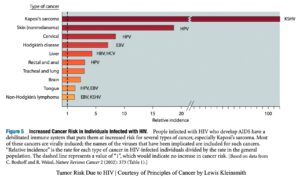
So why do these lesions occur on the skin’s surface? The skin is the largest organ in our body; therefore, we have a couple of different layers of skin. Within the skin, there are the epidermis and dermis. Blood vessels only reside in the dermis. Proteins derived from KSHV stimulate blood vessel growth in the dermis. It does so by combining KSHV-K1 mechanisms with HIV-1 proteins. Then, HIV-1 NEF proteins activate and stimulate cell proliferation.20 The number of blood vessels eventually increases as more channels form from previous vessels. There are other pathways involved in the development of KS from HIV/AIDS. Yet, it is important to note that the direct biomechanics for tumor conversion still need to be fully understood.
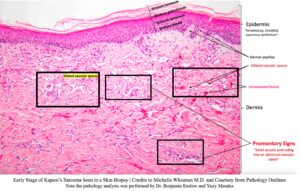
In the following image, a skin punch biopsy from an early case of KS is displayed. From the picture, we can see some key points. The first layer of the skin is the epidermis, an epithelial layer. The lowest sublayer of the epidermis is called the stratum basale, the layer of cells attached to the epidermis’s basement membrane. Normal cells cannot divide if they are not connected to a basal lamina or an extracellular matrix due to cell adhesion. On the contrary, cancerous cells can grow since they are not reliant on adhesion. The basal lamina and ECM support the cell as they are the foundation for the cellular layers. Notice the purple nuclei in the epidermis; that line indicates where the epithelial cells sit on the basal lamina. Also, the stratum basale contains stem cells, eventually becoming the stratum spinosum, granulosum, and corneum. In the stratum spinosum, the cells begin to flatten out and lose their cellular components to become fully packed with keratin. As the cells begin to flatten, hemidesmosome proteins, which aid in cell adhesion, will pull on the cells and make them appear spindle-shaped in the stratum granulosum and corneum. The stratum corneum is the exterior sublayer of the skin, the part we can feel. Since it contains no active organismal components like cells in deeper areas of the skin tissue, this sublayer serves as our protector. Keratin strengthens the skin by providing structural support to the body and protecting the internal organs from UV light damage and pathogen invasion.21
Just below the epidermis is the dermis. It features the dermal papillae, blood vessels, and extravasated blood. Since the epidermis has no blood supply, the dermis provides nutrients by diffusing them upwards into the finger-like structures (dermal papillae). The dermal papillae extend into the epidermis, allowing for the gas exchange of nutrients. A normal dermis layer contains blood vessels, but not as many as seen in the biopsy image. In the later stages of KS, the blood vessels undergo angiogenesis and extend toward the epidermis. The cells of the dermis and epidermis will flatten out completely (spindle cells), which will showcase the lumps from the growing vessels on the outer skin. At some point, the lumps become painful and may possibly even bleed when pressed against.
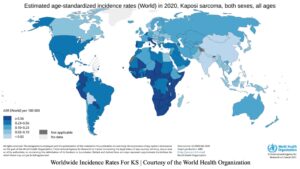
There are other variants of KS: classic, endemic (African), and transplant-related. In general, KS mainly affects individuals residing in the Middle East, Africa, and Eastern Europe.22, 23 It also affects the elderly due to weakened immune systems.24 In the late 1980s, once HIV/AIDS became more prevalent in the United States, a sudden increase in KS cases flooded the nation. By 1994, KSHV had been identified as being associated with KS. A study found that “…of 11,135 AIDS-related KS cases, 9356 were diagnosed from 1980 to 1995 and 1779 from 1996 to 2005…”.25 As cancer research progressed rapidly, a few treatments became available to the public, primarily those in first-world countries. At the moment, developing nations are most affected by KS because they lack the same access to healthcare as the U.S. and Europe have. If KS goes untreated, it will be deadly to the patient.
Currently, there are no treatments for KS since the available treatments have been unable to eradicate the HHV-8 infection.26 Few treatments are available to patients, but the most promising one at the moment is the Highly Active Antiretroviral Therapy (HAART). HAART is a treatment comprising a combination of retroviral drugs, nucleoside analog reverse transcriptase (RT) inhibitors, non-nucleoside analog RT inhibitors, and protease inhibitors.27,28,29 The RT inhibitors limit the ability of the KSHV to undergo reverse transcriptase, which is when a virus makes its DNA from its viral mRNA by infecting a host cell. Eventually, the host cell will divide with the viral DNA implemented into its genome because p53 will be mutated. As a result, p53 will lack its DNA-damaged or apoptosis-stimulating mechanism, allowing for cell proliferation.30, 31 T he KSHV virus has been found to play a role in p53 protein deactivation; however, its pathway is not fully understood.32
he KSHV virus has been found to play a role in p53 protein deactivation; however, its pathway is not fully understood.32
The HAART treatment is used when large quantities of the virus are found in an individual and restores CD4 T-cell counts.33,34 CD4 T lymphocyte cells stimulate other immune cells to target a pathogen and fight against it. When there’s a low level of CD4 T-cell activity, the immune system cannot recognize and combat pathogens. This combination of treatments has reduced patient complications and restored pathogen immunity.35
HIV is a very complex virus that constantly mutates and halts the immune system’s response. Although there’s treatment that aids in diminishing the symptoms of HIV or HIV/AIDS, it’s not a cure for patients. Currently, promising results have been observed with HAART treatment as patients’ KS symptoms alleviate efficiently and disappear from patients.36 Other treatments undergoing clinical experimentation are MMP protein inhibitors and genetically engineered virus lysis. Hindering MMPs would limit cancer’s ability to metastasize, thereby limiting its aggression on the body. MMP inhibitors may be used in multiple cancers since they target their degrading properties. There are attempts to develop a treatment that engineers a virus to target and destroy cancerous cells.37 The timeline for this is unknown since drug discovery can take somewhere between 10 to 15 years, but some engineered viruses have been successful in limiting metastasis and tumor invasion. Hopefully, in the coming generations, we will find a universal cure for cancer. Finding one cure will help the science and medical community better understand other cancerous properties.
- What is cancer? – Nci. (2007, September 17). https://www.cancer.gov/about-cancer/understanding/what-is-cancer. ↵
- Alberts, B., Johnson, A., Lewis, J., Raff, M., Roberts, K., & Walter, P. (2002). The initiation and completion of DNA replication in chromosomes. In Molecular Biology of the Cell. 4th edition. Garland Science. https://www.ncbi.nlm.nih.gov/books/NBK26826/ ↵
- How do cells divide?: MedlinePlus Genetics. (n.d.). Retrieved October 20, 2023, from https://medlineplus.gov/genetics/understanding/howgeneswork/cellsdivide/ ↵
- G1 and g2: What happens in the growth phases of the cell cycle? (2019, December 18). Albert Resources. https://www.albert.io/blog/g1-g2-phases-cell-cycle/ ↵
- Caretaker gene—An overview | sciencedirect topics. (n.d.). Retrieved October 20, 2023, from https://www.sciencedirect.com/topics/biochemistry-genetics-and-molecular-biology/caretaker-gene ↵
- Classification & structure of blood vessels | seer training. (n.d.). Retrieved October 20, 2023, from https://training.seer.cancer.gov/anatomy/cardiovascular/blood/classification.html. ↵
- Kleinsmith, L. J. (2006). Principles of cancer biology. Pearson Benjamin Cummings. ↵
- Kleinsmith, L. J. (2006). Principles of cancer biology. Pearson Benjamin Cummings. ↵
- Kleinsmith, L. J. (2006). Principles of cancer biology. Pearson Benjamin Cummings. ↵
- Kleinsmith, L. J. (2006). Principles of cancer biology. Pearson Benjamin Cummings. ↵
- Kleinsmith, L. J. (2006). Principles of cancer biology. Pearson Benjamin Cummings. ↵
- Kleinsmith, L. J. (2006). Principles of cancer biology. Pearson Benjamin Cummings. ↵
- Diagnosed, J. (n.d.). Va. Gov | veterans affairs. Retrieved October 21, 2023, from https://www.hiv.va.gov/patient/diagnosis/OI-common-illnesses.asp. ↵
- Kleinsmith, L. J. (2006). Principles of cancer biology. Pearson Benjamin Cummings. ↵
- Kaposi sarcoma. (2023, September 28). https://www.hopkinsmedicine.org/health/conditions-and-diseases/sarcoma/kaposi-sarcoma. ↵
- Kleinsmith, L. J. (2006). Principles of cancer biology. Pearson Benjamin Cummings. ↵
- Kaposi sarcoma. (2023, September 28). https://www.hopkinsmedicine.org/health/conditions-and-diseases/sarcoma/kaposi-sarcoma. ↵
- Kleinsmith, L. J. (2006). Principles of cancer biology. Pearson Benjamin Cummings. ↵
- Zeng, Z., Guan, L., An, P., Sun, S., O’Brien, S. J., Winkler, C. A., & the HBV study consortium. (2008). A population-based study to investigate host genetic factors associated with hepatitis B infection and pathogenesis in the Chinese population. BMC Infectious Diseases, 8(1), 1. https://doi.org/10.1186/1471-2334-8-1. ↵
- B;, B. P. J. P. (n.d.). P53 tumor suppressor protein stability and transcriptional activity are targeted by Kaposi’s sarcoma-associated herpesvirus-encoded viral interferon regulatory factor 3. Molecular and cellular biology. https://pubmed.ncbi.nlm.nih.gov/24248600/. ↵
- Keratin structure, function & diseases—Video & lesson transcript. (n.d.). Study.Com. Retrieved October 20, 2023, from https://study.com/WEB-INF/views/jsp/redesign/academy/lesson/seoLessonPage.jsp. ↵
- Kleinsmith, L. J. (2006). Principles of cancer biology. Pearson Benjamin Cummings. ↵
- Haart therapy drugs : Goals. YouTube. (2019, November 13). https://youtu.be/9qGwLhaBXeo?si=WpsQdLPwuGKj6UDB. ↵
- Haart therapy drugs : Goals. YouTube. (2019, November 13). https://youtu.be/9qGwLhaBXeo?si=WpsQdLPwuGKj6UDB. ↵
- Armstrong, A. W., Lam, K. H., & Chase, E. P. (2012, March 12). Epidemiology of classic and AIDS-related Kaposi’s sarcoma in the USA: Incidence, survival, and geographical distribution from 1975 to 2005: Epidemiology & Infection. Cambridge Core. https://doi.org/10.1017%2FS0950268812000325. ↵
- Kaposi sarcoma treatment & management: Approach considerations, medical care, local therapy. (2022). https://emedicine.medscape.com/article/279734-treatment?form=fpf. ↵
- Kleinsmith, L. J. (2006). Principles of cancer biology. Pearson Benjamin Cummings. ↵
- Martinez, V., Caumes, E., Gambotti, L., Ittah, H., Morini, J.-P., Deleuze, J., Gorin, I., Katlama, C., Bricaire, F., & Dupin, N. (2006). Remission from Kaposi’s sarcoma on HAART is associated with suppression of HIV replication and is independent of protease inhibitor therapy. British Journal of Cancer, 94(7), 1000–1006. https://doi.org/10.1038/sj.bjc.6603056. ↵
- Haart therapy drugs : Goals. YouTube. (2019a, November 13). https://youtu.be/9qGwLhaBXeo?si=WpsQdLPwuGKj6UDB. ↵
- B;, B. P. J. P. (n.d.). P53 tumor suppressor protein stability and transcriptional activity are targeted by Kaposi’s sarcoma-associated herpesvirus-encoded viral interferon regulatory factor 3. Molecular and cellular biology. https://pubmed.ncbi.nlm.nih.gov/24248600/. ↵
- Functional p53 signaling in Kaposi’s sarcoma-associated herpesvirus … (n.d.). https://journals.asm.org/doi/pdf/10.1128/JVI.01757-06. ↵
- Functional p53 signaling in Kaposi’s sarcoma-associated herpesvirus … (n.d.). https://journals.asm.org/doi/pdf/10.1128/JVI.01757-06. ↵
- Manes, T. D., Hoer, S., Muller, W. A., Lehner, P. J., & Pober, J. S. (2010). Kaposi’s sarcoma-associated herpesvirus K3 and K5 proteins block distinct steps in transendothelial migration of effector memory CD4+ T cells by targeting different endothelial proteins. Journal of immunology (Baltimore, Md. : 1950), 184(9), 5186–5192. https://doi.org/10.4049/jimmunol.0902938. ↵
- Program, aInfectious D. C. R. (n.d.). Is Kaposi’s sarcoma occurring at higher CD4 cell counts… : AIDS. LWW. https://journals.lww.com/aidsonline/pages/articleviewer.aspx?year=2010&issue=11270&article=00017&type=Fulltext. ↵
- Haart therapy drugs : Goals. YouTube. (2019, November 13). https://youtu.be/9qGwLhaBXeo?si=WpsQdLPwuGKj6UDB. ↵
- Kleinsmith, L. J. (2006). Principles of cancer biology. Pearson Benjamin Cummings. ↵
- Kleinsmith, L. J. (2006). Principles of cancer biology. Pearson Benjamin Cummings. ↵


2 comments
Jailene Ortiz
Great work! I learned a lot and especially when you started with “ we see that their health quickly deteriorates due to the ongoing physical symptoms, even with treatment”. I find it devastating knowing that people that are in chemo don’t really have that chance to 100% recover. Cancer is a huge deal and this kind of research is what we need!
Carlos Cenoz
Wow, this article was very well written. I learned so much about the mechanisms of cancer and its progression. It is very interesting to learn about these concepts and how they may affect our bodies. I also did not know that individuals with HIV/AIDS were prone to other diseases. Good job!