
In today’s world, it is common to hear news about a new drug targeting cancer or another disease. Many pharmaceutical drugs and treatments are approved for treating many diseases; however, have you ever stopped to think about the process that drug discovery takes? In scientific research, often animal subjects are used for drug development, but when does this transition to human subjects?
Clinical trials are defined by the National Institute of Health (NIH) as “…research studies that test a medical, surgical, or behavioral intervention in people.”.1 Clinical trials are conducted before introducing a new form of technology, whether it is a medical device or a new pharmaceutical drug, to become a treatment for the general public. Introducing and researching new tests and treatments is used to evaluate their effects on human health outcomes.2 Before starting a clinical trial, a few factors need to be listed since it is not always easy to get approved to conduct a trial. An application must be sent to the Food and Drug Administration (FDA), which will be evaluated by the Institutional Review Board (IRB), consisting of experts in the field. The application that is filed contains all the information regarding the trial. IRB approval is required for all human research, which consists of extensively evaluating methods and procedures, tests, or treatments of interest. This stage is referred to as the Pre-Clinical phase.3 The application must include a research question, hypothesis, tests or treatment of interest, target population, adequate bias control measures, justifications for the significance of the clinical trial(s), location and timeline, methods for data collection, budget planning, documentation of trained personnel, and sample recruitment.4 Applications are not subject to only the listed items. Each application differs; therefore, researchers must consider many other factors before submitting their application to the IRB. The primary role of the FDA is to ensure the rights and safety of human subjects are respected during clinical testing, evaluate the efficacy of new treatments, and compare the risks and benefits of new drugs to the ones on the market.5 These factors play a huge role in determining if treatments proceed to the next stage of the Clinical Phase.
After compiling set requirements and receiving approval from the IRB, treatment may proceed to the clinical trial. This section has three phases for pharmaceutical drug and/or treatment evaluation. The initial Clinical Stage, Phase 1, better understands the drug’s pharmacokinetics. Phase 1 Trial studies are performed to establish a treatment’s safe dose, decide how to administer it, and understand its effects on the body.5 7 8 Determining the highest dose that can be tolerated, the maximum tolerated dose, is the aim of this phase. Since information regarding the functions of the treatment is being collected, there is a very small sample size, which then gives faster results. Enrollment of participants falls somewhere between twenty to eighty patients This phase may last several months, and about 70% of pharmaceutical treatments proceed to the following phase.9 After establishing a safe dose, the treatment may move to Phase 2, including the Clinical Pharmacological Evaluation stage.
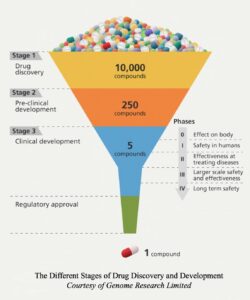
In this phase, the treatment is extensively evaluated as a potential new medication based on its efficacy for a specific disease or condition.7 11 The sample population for Phase 2 Trials is between one hundred and three hundred participants.9 11 Usually, randomized double-blinded trials are preferred. A randomized trial is when participants are randomly assigned to the experimental or control group. The experimental group will be given the treatment of interest, while the control will be given a standard treatment or a placebo. Double-masked studies refer to neither the researchers nor the subjects knowing who received what treatment. Double-blinded experiments limit bias and prevent the introduction of new ones.14 According to the FDA, Phase 2 trials can last a few months or up to two years, and approximately thirty-three percent of drugs move to the next phase.9 If the benefits of the treatment in this stage outweigh the risks, it will move on to Phase 3 Trials, where the extended clinical evaluation will occur. Phase 3 Trials are usually randomized and blinded studies that will have between 300 to 3,000 participants.9 17
The third phase compares the current treatment on the market with the proposed one. The results will determine if the proposed clinical trial treatment benefits outweigh the one on the market for the disease or outcome of interest. In Phase 3, about twenty to thirty percent of treatments move on to the next phase, which consists of the FDA reviewing all of the data collected in the Clinical Phase.5 19
Depending on the results of all three phases, a clinical trial may submit a New Drug Application (NDA) for FDA approval. This application will include information regarding the drug’s proposed labeling, patent information, directions for use, the IRB’s compliance information, information for drug abuse, and safety updates.20 If any research was conducted outside the United States, data from these studies must also be included. The FDA’s role is to confirm the drug’s safety and effectiveness after analyzing the collected data. If the FDA approves the target treatment, then it will proceed to Phase 4. More studies are conducted in this stage to learn about the treatments’ side and long-term effects.21 20 The number of enrolled participants is several thousand, so trials can last a few years. In summary, participating or starting a clinical trial is a very lengthy process and on average, regulatory steps for drug discovery and development take about ten to fifteen years.

Knowing what we know about clinical trials, how do we protect human subjects? Since the earliest of time, many medical and research practices did not consider the pillars of medical ethics, such as autonomy, beneficence, nonmaleficence, and justice of human subjects. From former primary education history classes, we came to learn about the harsh and unsanitary working conditions in the United States. Since there was a lack of knowledge regarding infectious disease, proper sanitary protocol, and slow biotechnological advances, many individuals suffered from severe illnesses and even death. In the early 1900s, there started to be an implementation of regulations that protected the autonomy of the general public. A changing point for protection was The Tuskegee Study of 1932 .23 This study was performed to study the effects of untreated syphilis on African-American men. The participants were told that they would be treated for “bad blood” and receive free medical treatment for their participation. Men with syphilis were withheld treatment when penicillin became available so that the researchers could continue monitoring the effects of the untreated disease. Many patients suffered organ damage and even death due to the lack of treatment.24 The study ended in 1972 after a press release explained the events of the study by a former participant. The Tuskegee Study led to the establishment of the National Commission for the Protection of Human Subjects of Biomedical and Behavioral Research in 1974.25
Historical events such as the Holocaust influenced the establishment of the Nuremberg Code in 1947, which contains a series of regulations required for research studies using human subjects that involve informed consent, experimentation should benefit society and have little to no harm to participants.26 The Declaration of Helsinki is more specific than the Nuremberg Code, which states the “…ethical principles for medical research involving human subjects, including research on identifiable human materials and data…”.27 Thirdly, The Belmont Report of 1978 states the respect of persons, beneficence, and justice.28 Many other policies and regulations have been placed to protect the rights of human subjects taking part in research.
Considering everything discussed and scientific advances, what does the future of clinical trials look like with the implementation of gene editing biotechnology? How will that affect societal norms? Recently, there has been a rise in interest in using biotechnology CRISPR-Cas9 (it is short for Clustered Regularly Interspaced Short Palindromic Repeats-associated protein 9). The CRISPR biotechnology was co-invented in 2012 by biochemist Jennifer Doudna and microbiologist Emmanuelle Charpentier.29 CRISPR-Cas9 has become the revolutionary discovery of our generation because it has provided a foundation for gene editing. It allows altering genomic sequences by programming CRISPR-Cas9 to target the gene of interest. Some of the main ethical concerns revolving around the use of CRISPR have been, “Is it ethical to alter our human genome?”. Many have argued that CRISPR biotechnology should not be used on humans because there is no research regarding human subjects. All of the information collected from this biotechnology has been due to animal subjects. One of the more prominent arguments is that humanity will play God and that altering genomic sequences will cause a new social order, a genetic hierarchy.
When the technology became commercially available, pharmaceutical companies and educational institutions began experimenting with it. Due to the extensive research, some institutions have shifted to collecting human subjects for clinical testing to determine whether CRISPR can target oncogenes or other disease-inducing genes.30 One of those institutions is the University of Minnesota (UMN), which has opened a Phase 2 clinical trial to treat metastatic gastrointestinal cancers in human subjects using CRISPR technology.31 With this clinical trial, UMN wants to delete the Cytokine-Induced SH2 (CISH) gene, a protein found in T-cells that helps the immune system fight against cancer. Deleting this gene would help T-cells recognize the cancer and find ways to eradicate it.21 CRISPR would eliminate the gene, targeting the gene of interest, the CISH gene on the T-cells. If CRISPR can do so, the T-cells can limit the growth of cancerous tumors within the body.
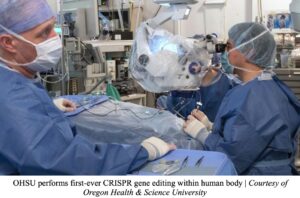 The University of Pennsylvania (UPenn) is another institution that treated two human subjects in a Phase 1 trial; one had multiple myeloma, a cancerous white blood cell formed in bone marrow, and the other had multiple sarcoma, a cancer affecting the bone marrow and/or soft tissue. Both of these individuals had relapsed, meaning they faced recurring cancers with standard treatments, and therefore sought to try CRISPR as a form of therapy.32
The University of Pennsylvania (UPenn) is another institution that treated two human subjects in a Phase 1 trial; one had multiple myeloma, a cancerous white blood cell formed in bone marrow, and the other had multiple sarcoma, a cancer affecting the bone marrow and/or soft tissue. Both of these individuals had relapsed, meaning they faced recurring cancers with standard treatments, and therefore sought to try CRISPR as a form of therapy.32
Access to such technology will benefit the medical community as disease treatment will be tailored to an individual. CRISPR could help find cures for incurable diseases and conditions that will be available to the public. Suffering individuals would be able to enroll in clinical trials or maybe even attend a doctor’s visit for a procedure using CRISPR biotechnology.
Now, CRISPR has shown promising results in multiple studies, but the long-term effects of using CRISPR have yet to be discovered due to its recent discovery. If more studies continue to show the benefits of this biotechnology, it will most likely become a societal norm. It will be used to cure diseases that do not have a cure, such as rare genetic conditions, metastatic cancers, and degenerative conditions due to age. However, we must ask, “In what scenario would it be ideal not to introduce CRISPR as a form of treatment? Will regulation (for protecting human subjects) adapt to including genetic testing? What are the long-term effects of genetic testing?” These are all questions one must consider when researching such revolutionary biotechnology.
Three independent studies have been conducted on the adverse effects of using CRISPR biotechnology in embryos: the “…unwanted heritable genomic changes…” such as DNA sequence deletions in chromosomes and CRISPR binding in incorrect DNA regions not targeting the gene of interest.33 Deleting sequences in the DNA can often cause an individual to become more susceptible to disease or even lead to death. For example, an embryo who faced issues while undergoing gene editing in the womb is more susceptible to body disfiguration, development of neurological disorders, and a shorter lifespan.34 35 36 The symptoms displayed will vary depending on the affected area for DNA deletions and the effect of using an abnormal CRISPR protein.

As mentioned before, CRISPR-Cas9 is the revolutionizing biotechnology of our generation. However, before undergoing any genetic editing, one must consider that participating in base editing would change the genetic linkage for future generations. The CRISPR protein is not guaranteed to work perfectly; therefore, complications may arise. It is essential to consider all possible scenarios, the good and bad, of taking part in genetic editing. Enrolling in a general clinical trial is a big decision, often the last resort for very sick patients. Now imagine enrolling in a genetic clinical trial; suppose unexpected issues were to arise during a procedure. In that case, it is expected that a patient and their families will face some sort of psychological stress as a response to the complications.37 38 Mental, physical, and emotional trauma could jeopardize the health of patients and their loved ones by making them more susceptible to disease. Sometimes, participating individuals’ socioeconomic and background factors often stimulate these symptoms.37 Lastly, one must consider that undergoing genetic editing may be irreversible. If complications arose, returning the genome to its original state would be very difficult, especially if the hope is to reverse the effects of CRISPR.
CRISPR-Cas9 has shown promising results. Much like other treatments, it is not always effective and needs to undergo extensive research to fully understand its editing capacity. Due to its recent discovery and controversy, only a few genetic clinical trials have been approved on human subjects, as many ethical concerns need to be considered. As we progress in genetic and molecular research, this biotechnology may become easily available within our generation. Then, would you participate in genetic clinical trials?
- National Institute on Aging. “What Are Clinical Trials and Studies?” Accessed September 29, 2023. https://www.nia.nih.gov/health/what-are-clinical-trials-and-studies. ↵
- Commissioner, Office of the. “Basics About Clinical Trials.” FDA, May 8, 2023. https://www.fda.gov/patients/clinical-trials-what-patients-need-know/basics-about-clinical-trials. ↵
- Drugs.com. “FDA Drug Approval Process.” Accessed September 29, 2023. https://www.drugs.com/fda-approval-process.html. ↵
- brandon.haslip@castoredc.com. “10 Things to Consider Before Starting Your Clinical Research Study.” Castor, November 12, 2018. https://www.castoredc.com/blog/starting-clinical-study/. ↵
- Hanson, Glen, Peter J. Venturelli, and Annette E. Fleckenstein. Drugs and Society. Thirteenth edition. Burlington, MA: Jones & Bartlett Learning, 2018. ↵
- Hanson, Glen, Peter J. Venturelli, and Annette E. Fleckenstein. Drugs and Society. Thirteenth edition. Burlington, MA: Jones & Bartlett Learning, 2018. ↵
- “Trial Phases 1, 2 & 3 Defined.” Accessed September 29, 2023. https://med.uc.edu/depart/psychiatry/research/clinical-research/crm/trial-phases-1-2-3-defined#:~:text=Phase%20II,approximately%20100%20to%20300%20volunteers. ↵
- What Are Clinical Trial Phases? Accessed September 29, 2023. https://www.youtube.com/watch?v=dsfPOpE-GEs. ↵
- Commissioner, Office of the. “Step 3: Clinical Research.” FDA, April 18, 2019. https://www.fda.gov/patients/drug-development-process/step-3-clinical-research. ↵
- “Trial Phases 1, 2 & 3 Defined.” Accessed September 29, 2023. https://med.uc.edu/depart/psychiatry/research/clinical-research/crm/trial-phases-1-2-3-defined#:~:text=Phase%20II,approximately%20100%20to%20300%20volunteers. ↵
- What Are Clinical Trial Phases? Accessed September 29, 2023. https://www.youtube.com/watch?v=dsfPOpE-GEs. ↵
- Commissioner, Office of the. “Step 3: Clinical Research.” FDA, April 18, 2019. https://www.fda.gov/patients/drug-development-process/step-3-clinical-research. ↵
- What Are Clinical Trial Phases? Accessed September 29, 2023. https://www.youtube.com/watch?v=dsfPOpE-GEs. ↵
- Gordis, Leon. Epidemiology. Fifth edition. Philadelphia, PA: Elsevier/Saunders, 2014. ↵
- Commissioner, Office of the. “Step 3: Clinical Research.” FDA, April 18, 2019. https://www.fda.gov/patients/drug-development-process/step-3-clinical-research. ↵
- Commissioner, Office of the. “Step 3: Clinical Research.” FDA, April 18, 2019. https://www.fda.gov/patients/drug-development-process/step-3-clinical-research. ↵
- “Trial Phases 1, 2 & 3 Defined.” Accessed September 29, 2023. https://med.uc.edu/depart/psychiatry/research/clinical-research/crm/trial-phases-1-2-3-defined#:~:text=Phase%20II,approximately%20100%20to%20300%20volunteers. ↵
- Hanson, Glen, Peter J. Venturelli, and Annette E. Fleckenstein. Drugs and Society. Thirteenth edition. Burlington, MA: Jones & Bartlett Learning, 2018. ↵
- Commissioner, Office of the. “Step 3: Clinical Research.” FDA, April 18, 2019. https://www.fda.gov/patients/drug-development-process/step-3-clinical-research. ↵
- MD Anderson Cancer Center. “Phases of Clinical Trials.” Accessed September 30, 2023. https://www.mdanderson.org/patients-family/diagnosis-treatment/clinical-trials/phases-of-clinical-trials.html. ↵
- Commissioner, Office of the. “Step 4: FDA Drug Review.” FDA, April 18, 2019. https://www.fda.gov/patients/drug-development-process/step-4-fda-drug-review. ↵
- MD Anderson Cancer Center. “Phases of Clinical Trials.” Accessed September 30, 2023. https://www.mdanderson.org/patients-family/diagnosis-treatment/clinical-trials/phases-of-clinical-trials.html. ↵
- “Tuskegee Study – Research Implications – CDC – OS,” December 20, 2022. https://www.cdc.gov/tuskegee/after.htm. ↵
- The Tuskegee Study. Accessed September 30, 2023. https://www.youtube.com/watch?v=afwK2CVpc9E. ↵
- “Tuskegee Study – Research Implications – CDC – OS,” December 20, 2022. https://www.cdc.gov/tuskegee/after.htm. ↵
- Shuster, Evelyne. “Fifty Years Later: The Significance of the Nuremberg Code.” New England Journal of Medicine 337, no. 20 (November 13, 1997): 1436–40. https://doi.org/10.1056/NEJM199711133372006. ↵
- “WMA – The World Medical Association-WMA Declaration of Helsinki – Ethical Principles for Medical Research Involving Human Subjects.” Accessed September 30, 2023. https://www.wma.net/policies-post/wma-declaration-of-helsinki-ethical-principles-for-medical-research-involving-human-subjects/. ↵
- Protections (OHRP), Office for Human Research. “Read the Belmont Report.” Text. HHS.gov, January 15, 2018. https://www.hhs.gov/ohrp/regulations-and-policy/belmont-report/read-the-belmont-report/index.html. ↵
- “Jennifer Doudna: National Inventors Hall of Fame.” The National Inventors Hall of Fame. Accessed November 2, 2023. https://www.invent.org/inductees/jennifer-doudna#:~:text=Biochemist%20Jennifer%20Doudna%20and%20microbiologist,across%20medicine%2C%20biology%20and%20agriculture. ↵
- White, Franny. “OHSU Performs First-Ever CRISPR Gene Editing within Human Body.” OHSU News, March 4, 2020. https://news.ohsu.edu/2020/03/04/ohsu-performs-first-ever-crispr-gene-editing-within-human-body. ↵
- University of Minnesota. “University of Minnesota Opens First of Its Kind Clinical Trial to Treat Metastatic GI Cancers Using CRISPR Genetic Engineering,” October 12, 2020. https://twin-cities.umn.edu/news-events/university-minnesota-opens-first-its-kind-clinical-trial-treat-metastatic-gi-cancers. ↵
- Stein, Rob. “First U.S. Patients Treated with CRISPR as Human Gene-Editing Trials Get Underway.” NPR, April 16, 2019. https://www.npr.org/sections/health-shots/2019/04/16/712402435/first-u-s-patients-treated-with-crispr-as-gene-editing-human-trials-get-underway. ↵
- Frosch, Jennifer. “Genome Editing in Human Embryos Has Unintended Side-Effects • Pet.” PET, September 28, 2022. https://www.progress.org.uk/genome-editing-in-human-embryos-has-unintended-side-effects/. ↵
- Alanis-Lobato, Gregorio, Jasmin Zohren, Afshan McCarthy, Norah M.E. Fogarty, Nada Kubikova, Emily Hardman, Maria Greco, Dagan Wells, James M.A. Turner, and Kathy K. Niakan. “Frequent Loss-of-Heterozygosity in CRISPR-Cas9-Edited Early Human Embryos.” bioRxiv, January 1, 2020. https://www.biorxiv.org/content/10.1101/2020.06.05.135913v1. ↵
- Author links open overlay panelMichael V. Zuccaro 1 2 8, 1, 2, 8, 3, 4, 5, et al. “Allele-Specific Chromosome Removal after Cas9 Cleavage in Human Embryos.” Cell, October 29, 2020. https://www.sciencedirect.com/science/article/pii/S0092867420313891?via%3Dihub. ↵
- Frequent gene conversion in human embryos induced by double … – biorxiv. Accessed November 2, 2023. https://www.biorxiv.org/content/10.1101/2020.06.19.162214v1. ↵
- Risk and emotion among healthy volunteers in clinical trials. Accessed November 2, 2023. https://journals.sagepub.com/doi/10.1177/0190272516657655. ↵
- “Psychological and Emotional Impact in Patients Undergoing Treatment For Metastatic Cancer Either in a Clinical Trial or as Standard Off-Trial Therapy.” CTG Labs – NCBI. Accessed November 2, 2023. https://clinicaltrials.gov/study/NCT00896467. ↵
- Risk and emotion among healthy volunteers in clinical trials. Accessed November 2, 2023. https://journals.sagepub.com/doi/10.1177/0190272516657655. ↵


1 comment
Jailene Ortiz
Wow! I found this research impressive! It’s intimidating to know that there’s a vaccine that can cure diseases but can only go into intervention with humans instead of animals.