
Silent Threat: The Chilling Reality of Creutzfeldt-Jakob Disease
Introduction
The neurodegenerative condition “Creutzfeldt-Jakob Disease” (CJD) is fatal in animals and humans.1 Ataxia, dementia, and other neurological symptoms manifest rapidly in those suffering from this condition. CJD is brought on by misfolding a normally occurring prion protein into a prion form. An abnormal isoform of prion protein (PrP(Sc)) builds up in the brain, causing neuronal death and the onset of CJD symptoms. The precise processes by which PrP(Sc) leads to neurodegeneration remain unclear. However, the fundamental event in the illness process is the misfolding of PrP into PrP(Sc). Researchers have hypothesized that PrP(Sc) is either directly harmful to neurons or indirectly toxic by interfering with normal cellular function, which would lead to neurodegeneration.
The pathogenesis of CJD is a multistep process involving the collection of PrP(Sc) in the brain, its misfolding into PrP(Sc), and the subsequent neurodegeneration. It explains the importance of these results for discovering new treatments for CJD and evaluates the evidence supporting several suggested mechanisms of neurodegeneration in this disease.
Background of the Disease
The neurodegenerative disease, Creutzfeldt-Jakob disease (CJD), is severe in humans and other animals.2 The four categories recognized to date are familial, variant, iatrogenic, and sporadic CJD. Most occurrences occur between the ages of 60 and 80; the disease has a short duration and starts at the average age of 65 years.3 It is theorized to arise from the propagation of a mutated form of a protein molecule known as prion protein. Cognitive impairment is the most common early indication of this disease, occurring in up to 35 percent of patients. In approximately 17.5% of reports, the first symptom may be cerebellar dysfunction, behavioral signs, or symptoms. About 85 percent of cases of CJD are due to the sporadic type of the disease.4
The median survival period is 3.5-6 months with cases of 1-1.5 per million, sporadic CJD is a severe neurological disease with unknown cause. Over weeks or months, it causes a gradual decline in cognitive abilities, coordination, and social behavior. An abnormal increase of prion protein in the neurons is responsible for the spongiform change, neuronal loss, and gliosis that characterizes CJD.
CJD symptoms can manifest in many ways, physically and mentally making it challenging to diagnose. Clinical presentation, examination, and imaging procedures, including EEG, MRI, and cerebrospinal fluid (CSF), regularly recommend CJD. However, a neuropathologic and immunodiagnostic examination of brain tissue acquired from a biopsy or autopsy is required to diagnose CJD accurately.
Causes, Transmission, and Incidence
Diseases can arise sporadically by inheritance or transmission. Sporadic infection is more common in humans, but research has focused primarily on contagious diseases. Ingestion, blood transfusion, and iatrogenic transmission are ways this disease can spread between species. Because the pathogenic agent (a prion) can remain in the atmosphere for several years, environmental exposure as a means of transmission cannot be discounted. Transformation of the prion protein (PrPC) to scrapie prion protein (PrPSc) causes neurodegenerative prion diseases.5 PrPSc, the primary component of prion, functions as an infectious template that changes normal cellular PrPC into its pathological, misfolded version. Several other disease-related proteins, including amyloid (A), tau, and -synuclein, have exhibited prionoid, or prion-like, spread. The recently revealed physiological roles of prion protein and the improved understanding of the etiology of prion disease. Prion protein has been linked to developing several neurological disorders besides prion disease. Prion disease therapeutic research has also used innovative ways of fighting these fatal illnesses.6
As two genetic investigations with mice suggested, infectious particles are likely formed from the natural PrPC protein. In one study, transgenic mice producing PrP with an alternative identical to that in GSS victims developed prion disease spontaneously. When transplanted into transgenic animals expressing modest quantities of similar mutant PrP protein, which are unlikely to develop the disease, the brain tissue from these animals can transfer prion disease. All elements required for pathogenic organisms appear endogenously present in the mice. It seems that the presence of wild-type PrP interfered in some way with disease development from the mutant transgene, as evidenced by the fact that eradication of the indigenous PrP gene in the most recent research resulted in earlier start of illness and more acute pathologies in the uninoculated transgene strain. A protein-only infectious agent causes structural changes in other proteins to spread the disease, making prion diseases a distinct class of infectious neurodegenerative disorders.7 Extensive and chronic brain deterioration is the cause of the condition. Neuronal death, Synaptic alterations, Spongiform degeneration, brain inflammation, and the accumulation of protein aggregates are all processes implicated in neurodegeneration, but our understanding of the molecular mechanisms involved is limited.
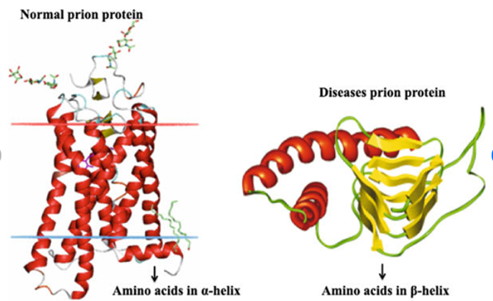
The most studied facet of prion disorders has been the unconventional character of an infectious agent. Protein, a conformational variant found in the cell prion protein located in the lymphatic and brain tissues, appears to be the predominant component of prions.8 The misfolded prion protein is distinct from normal prion protein since it contains a more significant proportion of aggregates and sheet structure to form polymers of intermediate and big sizes. The critical issue in prion illness is the transformation of normal to misfolded prion protein, but how it leads to disease and brain damage is unknown.
Clinical Presentation
Prion disorders are characterized by developing PrP(Sc) plaques in the brain. Neurons exposed to PrP(Sc) plaques may die off due to toxicity and neurodegeneration. PrP(Sc) buildup causes ER stress, which triggers the UPR as a protective mechanism against protein misfolding by upregulating different chaperones and reducing total protein expression. Neuronal death and maybe spongiform degeneration can be directly caused by prolonged ER stress. PrP(Sc) plaques have been hypothesized as a method by which neuronal function is altered. The regular signaling systems crucial to neuronal function can be disrupted when PrP(Sc) plaques bind to other proteins on the surface of neurons. Plaques made of PrP(Sc) have been hypothesized to play a role in this process by stimulating the creation of toxic substances. Plaques made of PrP(Sc) have been shown to stimulate the immune system. This can trigger the release of inflammatory substances that damage neurons. Blood-brain barrier disruption is another way in which PrP(Sc) plaques can cause damage to the brain. The brain is protected from potentially dangerous substances in the blood by a specialized network of cells known as the blood-brain barrier. PrP(Sc) plaques can compromise the blood-brain barrier, allowing toxic molecules to enter the brain, where they can kill neurons.
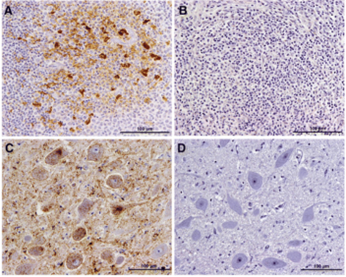
Patients with prion disease who have survived for several years typically display cerebral shrinkage similar to that seen in other neurodegenerative illnesses. However, no noticeable physical abnormalities exist in the brain. Neuropil vacuolation (a well-recognized light microscopic histological feature of many diverse diseases) in the gray matter, significant neuronal loss, synaptic abnormalities, exuberant reactive astrogliosis (morphological and functional changes seen in astroglial cells/astrocytes responding to CNS injury and other neurological diseases), and a varied extent of the cerebral buildup of prion protein accumulate all common neuropathological alterations associated with prion disorders. It is unclear how much these alterations contribute to brain illness and functioning. Here, we detail what is known about how these neurodegenerative alterations occur and their potential role in the disease’s pathophysiology. New treatment targets for prion illnesses could be found if the molecular basis of brain injury were understood.
Current Research
There is substantial proof that PrP(Sc) contributes to neurodegeneration. In CJD, PrP(Sc) plaques are located in the brain regions most impacted by neurodegeneration. Furthermore, PrP(Sc) is neurotoxic in laboratory experiments. It is known that PrP(Sc) is the pathogen behind prion diseases, but it is not clear how it causes neurodegeneration. Several mechanisms have been hypothesized, but none have been proven. PrP(Sc) has been hypothesized to cause direct damage to neurons. Neuronal membranes can develop pores when exposed to PrP(Sc), allowing ions and other molecules to enter the cell.9 The neuron’s normal function may be impaired and die as a result.
The synthesis of harmful chemicals in response to PrP(Sc) has also been postulated as a mechanism. PrP(Sc) in the brain has been linked to immune system activation. This can trigger the release of inflammatory chemicals that harm nerve cells. In addition to damaging neurons directly, PrP(Sc) can enter the brain and cause damage by disrupting the blood-brain barrier. The brain is protected from potentially dangerous substances in the blood by a specialized network of cells known as the blood-brain barrier. The blood-brain barrier can be compromised by PrP(Sc), allowing potentially neurotoxic chemicals to cross into the brain and do their damage there.10
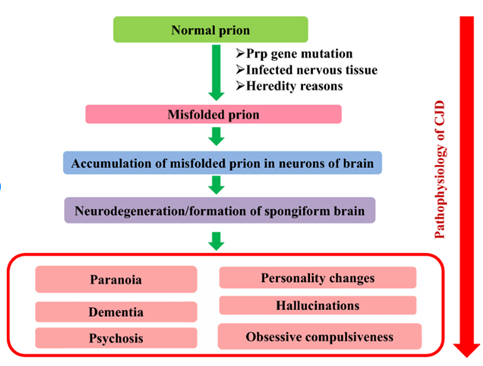
Furthermore, in 1982, Prusiner made the groundbreaking discovery that proteinaceous infectious agents exist. The presence of nucleic acids ensured the reproduction and survival of all known pathogens. Comparatively, prion proteins (PrP) were proteinaceous disease agents that did not contain nucleic acids and spread by binding to the practical, endogenous variant of cell prion protein (called PrPC) and changing its shape to produce the pathogenic, misfolded protein (called PrPSc). When these infectious prion proteins accumulate and aggregate in the brain, they destroy neural tissue and ultimately cause fatal spongiform encephalopathies.11 These discoveries are especially significant because the concepts of prion pathogenesis are also relevant to the pathogenesis of other neurodegenerative illnesses like Parkinson’s, amyotrophic lateral sclerosis, Huntington’s, and Alzheimer’s. The worldwide spread of these illnesses is on the rise, yet viable treatments are still unavailable.
Conclusion
Prion illnesses, including CJD, are multifaceted neurodegenerative conditions with complicated underlying molecular and cellular pathways. Research suggests that autophagy and apoptosis are responsible for neuronal death in prion disorders. Given their association with protein structural changes and relevance to understanding more considerable neurodegenerative illnesses, these diseases are of significant scientific interest. In addition, CJD and other prion diseases are not unique; they are linked to other neurodegenerative conditions via common genetic and clinical mechanisms.
The advancement of understanding neurodegeneration and the development of possible therapeutics depends on our ability to comprehend the complex mechanisms of prion disorders. In light of the complexity of these severe diseases, continuing study into their molecular and cellular features is essential. The precise processes by which PrP(Sc) induces neurodegeneration are still to be determined, but research is ongoing. New treatments for prion illnesses require a more profound knowledge of these mechanisms.
- Nathan R Deleault et al., Formation of Native Prions from Minimal Components in Vitro. Proceedings of the National Academy of Sciences of the United States of America, June 5, 2007, https://www.ncbi.nlm.nih.gov/pmc/articles/PMC1887554/. ↵
- Adrian Aguzzi and Mathias Heikenwalder.Pathogenesis of Prion Diseases: Current Status and Future Outlook. Nature Reviews. Microbiology, 2006, https://pubmed.ncbi.nlm.nih.gov/16980938/. ↵
- SB Prusiner.Prions. Proceedings of the National Academy of Sciences of the United States of America, 1998, https://pubmed.ncbi.nlm.nih.gov/9811807/. ↵
- SB Prusiner.Prions. Proceedings of the National Academy of Sciences of the United States of America, 1998, https://pubmed.ncbi.nlm.nih.gov/9811807/. ↵
- Claudio Soto and Nikunj Satani.The Intricate Mechanisms of Neurodegeneration in Prion Diseases. Trends in Molecular Medicine 17, no. 1 (2011): 14–24, https://doi.org/10.1016/j.molmed.2010.09.001. ↵
- Claudio Soto and Nikunj Satani. The Intricate Mechanisms of Neurodegeneration in Prion Diseases. Trends in Molecular Medicine 17, no. 1 (2011): 14–24, https://doi.org/10.1016/j.molmed.2010.09.001. ↵
- Caihong Zhu and Adriano Aguzzi. Prion Protein and Prion Disease at a Glance. Journal of Cell Science 134, no. 17 (2021), https://doi.org/10.1242/jcs.245605. ↵
- John Collinge. Neurotrophins: Roles in Neuronal Development and Function. Annual Reviews, 2001, https://www.annualreviews.org/doi/abs/10.1146%2Fannurev.neuro.24.1.677. ↵
- Arthur L Horwich and Jonathan S Weissman. Deadly Conformations—Protein Misfolding in Prion Disease. Cell 89, no. 4 (1997): 499–510, https://doi.org/10.1016/s0092-8674(00)80232-9. ↵
- Claudio Soto and Nikunj Satani. The Intricate Mechanisms of Neurodegeneration in Prion Diseases. Trends in Molecular Medicine 17, no. 1 (2011): 14–24, https://doi.org/10.1016/j.molmed.2010.09.001. ↵
- SB Prusiner. Prions. Proceedings of the National Academy of Sciences of the United States of America, 1998, https://pubmed.ncbi.nlm.nih.gov/9811807/. ↵

1 comment
aramirez104
I have always been fascinated by prion-related diseases and their development in the nervous system. It is really unfortunate that such a large population of CJD patients have sporadic onsets of this disease, and that overall patients with CJD are burdened with a survival rate in that range. I understand there has yet to be an answer for why prion protein misfolding affects the brain and results in diseases specific to the nervous system, but I hope that advancements in technology and further research find these answers for us in the future and can aid in treatment and increasing the life span for this fatal disorder.