
Introduction:
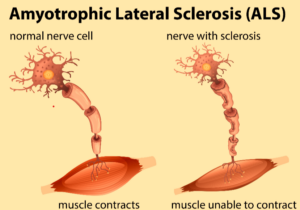
ALS, or Amyotrophic Lateral Sclerosis, is a neurodegenerative disease that causes loss of the upper and lower motor neurons in the body, which eventually leads to muscle wasting and an inability to move.1 The onset of ALS usually begins in late adulthood, around 50 to 75 years old, and is more prominent in white Caucasian males, although it can begin at any age, sex, race, or ethnicity. If the disease begins before the age of 25, it is considered juvenile ALS and only affects about 5% of patients who acquire the disease in their lifetime. There is currently no cure available for ALS, only medications that can slow the progression, which is why ALS is considered a terminal illness; most people only live 3 to 5 years after receiving a diagnosis.2 There are two types of ALS: sporadic and familial. Sporadic ALS accounts for 90% of all cases of ALS, and there is no genetic or hereditary risk involved. There are many reasons someone could acquire ALS sporadically, but some of the reasons include environmental factors such as toxins, a person’s lifestyle, viruses, and other diseases that cause ALS to begin. Familial ALS accounts for 10% of all cases of ALS, and there are genetic factors involved to cause the disease.3
Many mutations are known to cause familial ALS, and they include SOD1, C9orf72, TARDBP, and FUS. For familial ALS, not all these mutations have 100% penetrance and will not always cause ALS if the mutation is present and must have other factors present to cause ALS. One mutation though has 100% penetrance and does not need other outside factors to cause ALS; it is the fused in sarcoma mutation in exon 14 of chromosome number 16, c.1483 C>T, p.arg495 STOP.4 This mutation is unique because the mechanism of the disease is different than in other mutations, it can be difficult to diagnose, and there are few options for any type of treatment.
Mechanism Of Disease in C.1483 C>T P. arg495STOP in FUS ALS:
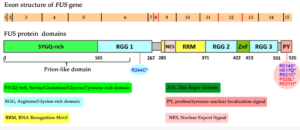
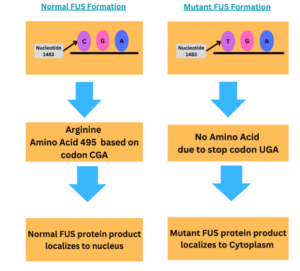
The mechanism of this disease for c.1483 C>T, p.arg495 STOP FUS ALS is not known for sure, but there is lots of research going on to find out how this mutation causes ALS. The FUS gene is 526 amino acids long, 15 exons long, and is a DNA and RNA-binding protein that is involved in gene regulation, gene transcription, mRNA metabolism, and DNA repair.6 In this specific mutation, the FUS protein is truncated, or shortened, by 31 amino acids early by a nonsense mutation. Cytosine is replaced by thymine on nucleotide 1483 on chromosome 16, exon 14 This change causes the stop codon (UAG), instead of coding for arginine (CAG). This then causes the protein to lose its nuclear localization signal and aggregate in the cytosol instead of advancing to the nucleus.7
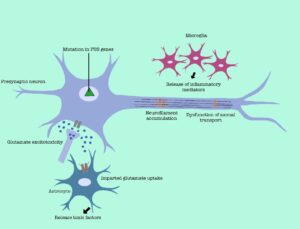
One proposed idea of how this mutation causes ALS is the accumulation of abnormal FUS in the cytosol that can cause excess stress granules to form and promote unregulated cellular processes such as excess inflammation that damages neurons, eventually causing neurodegeneration that leads to ALS. Stress granules are a combination of mRNA and misfolded proteins that aggregate in the cytosol when the cell is exposed to intracellular stress or extracellular stress, such as mutant proteins that are toxic to the cell, to ensure the cell’s survival. As soon as the stress dissipates from the cell, stress granules are supposed to disassemble and then dissolve. Still, with truncated FUS ALS mutations, stress granules do not disassemble in the cell, causing more toxicity to the cell.8 Another idea of how this mutation can cause ALS is by loss of function of FUS in the nucleus. Since mutant FUS aggregates in the cytoplasm, because the protein loses its nuclear localization signal, there is not enough normal FUS available in the nucleus to regulate cellular processes such as transcription, mRNA metabolism, DNA repair, and gene regulation. These processes end up failing because the one good allele that produces normal FUS is not enough to regulate these processes alone in the neurons, in turn causing the death of motor neurons to prevent more damage to the DNA. Since motor neurons cannot be regenerated after death, this ends up causing ALS because the connection at the neuromuscular junction is lost due to the death of the neuron, and the muscles and neurons are therefore unable to communicate with each other. 9
Two other theories of how this mutation can cause ALS is by the protein ubiquitin. The first theory is that mutant FUS in the cytoplasm is tagged with ubiquitin to be degraded by the proteasome, but since there is such a large amount of FUS in the cytoplasm that accumulates, the proteasome eventually reaches a threshold where no more mutant FUS can be degraded. The mutant FUS then aggregates in the cytoplasm, becomes toxic, and interferes with the connection between other neurons and the neuromuscular junction, eventually causing the death of motor neurons and the wasting of muscles, which causes paralysis. This is a reason why symptoms of ALS for this specific mutation arise at different ages in a person’s lifetime because it is not until the proteasome cannot degrade this mutant protein anymore that it becomes toxic and causes symptoms of ALS.10 The second theory of how ubiquitin can cause symptoms of ALS in this mutation is that since FUS is mutated ubiquitin cannot effectively bind to the protein and cannot prevent the FUS protein from transforming into its liquid phase, once the protein is in the liquid phase, it promotes more stress granules to form in cell thereby causing excess inflammation which leads to neuronal death and symptoms of ALS.11. There are many proposals as to why this mutation causes ALS, but it is likely a mix of all or some of these researched theories and possibly age, race, sex, and ethnicity dependent.
Structure of the FUS gene:
Normal and Mutant FUS Formation Flowcharts:
Different Mechanisms of How ALS Causes Neurodegeneration:
Difficulty of Diagnosing ALS:
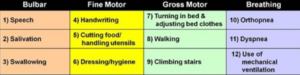
Diagnosis of ALS can be a difficult process because the disease can look similar to many other illnesses, and neurologists usually have to rule out all of these other illnesses before even considering ALS as a possibility. Sometimes even if neurologists consider ALS as a possible cause of the patients’ symptoms, they still have to perform many tests to confirm this. These tests include electromyography (EMG), magnetic resonance imaging (MRI), nerve conduction tests, muscle and nerve biopsies, blood and urine tests, genetic testing, and questionnaires to ask the patient about their functioning.13 One important questionnaire about the patient’s functioning is called the ALSFRS-R or ALS functional rating scale revised, this scale has a total of 12 questions, and each question asks about the person’s ability to perform a certain task, each question runs on a scale from 0 to 4, with 0 meaning the patient cannot perform a task, and 4 meaning the patient has full ability to perform a task. ALSFRS- R score is not only used for diagnostic processes but also for the patient after they receive a diagnosis to monitor their progression.14 This process for diagnosis on average can take anywhere from a year to three years to receive a diagnosis, and by this time the patients have usually already progressed far into the disease.15 For this mutation of ALS, diagnosis time varies greatly because it is a familial form of ALS, and not all patients are knowledgeable that they have a chance of acquiring this mutation and developing ALS. After all, genetic testing was not always readily available, as it is today, so not all their parents who had this mutation got tested. If the patient and neurologist are knowledgeable about the patient’s family history and the mutation, diagnosis time is typically faster, than if the patient and neurologist do not understand this specific mutation or the family history.
There are three types of symptom onset, and they are limb onset, bulbar onset, and mixed onset. Limb onset symptoms include muscle weakness in figures, toes, arms, and legs, tripping or falling, muscle twitches, muscle tremors, loss of muscle or muscle atrophy, stiff muscles, and cramping.16 Bulbar onset symptoms include trouble with swallowing, speech problems, tongue fasciculations, twitching of muscles in the face, trouble moving facial muscles, difficulty with breathing, and pseudobulbar affect which is the inability to control laughing or crying. Mixed onset is a combination of limb and bulbar onset symptoms.17,18 Patients who are diagnosed with bulbar onset ALS typically live less time about 10 months to 2 years after receiving a diagnosis than patients who are diagnosed with limb onset ALS, which live 2 to 5 years after a diagnosis is received. Patients who are diagnosed with mixed-onset ALS have a varied lifespan after receiving a diagnosis depending on the severity of the patient’s symptoms.19,20 Patients with this specific mutation can present with any symptom onset, but the ones that present with bulbar symptom onset typically do not live more than two years.21 This mutation, in general, is a very fast-progressing and severe form of ALS, most patients are diagnosed before the age of 40, and some of these patients are even diagnosed before the age of 25, this is known as juvenile ALS or jALS.22
Diagnosing this form of ALS comes with many challenges due to the presentation of the disease, the lack of knowledge of patients and neurologists about this mutation and the role it plays in ALS, the extremely rapid progression of this disease with this mutation, and the age range that ALS will present that this mutation.
ALSFRS-R Score Table:
Treatments For ALS:
There is no cure for ALS as of right now, but there are a few medications that can slow the progression of the disease depending on how early a patient starts medication, the type of onset of symptoms of the patient, and other factors as well. Rizoule is one medication that can delay the progression of ALS, by blocking the release of excess glutamate, since glutamate can damage neurons.24 Radicava is another drug approved by the FDA, and it works by reducing oxidative stress, which is a possible contributor to ALS.25 A 3rd medication approved by the FDA recently in 2022 is Relyvrio, which works by blocking stress signals in the endoplasmic reticulum (ER) and the mitochondria; stress signals increase neuronal damage, so this drug helps reduce it.26 These medications are not studied enough for this specific mutation to see if they work sufficiently, and the reason is that this mutation is so rare.
There are also clinical trials that patients can do if they meet the criteria to join, but for this mutation and other FUS mutations as well, there is currently only one trial that is currently available, and it is from the pharmaceutical company IONIS. The medication’s name is Ulefnersen or Jacifusen (previously known as ION 363), and it is predicted that this medication works by reducing mutant FUS by using antisense oligonucleotide therapy. Antisense oligonucleotide therapy in general works by inhibiting the translation of the mRNA into the target protein. Ulefnersen was first used in mice to see what it would do, and it ended up reducing mutant FUS as it was intended to do, but it worked best in the mice that were still in the mother’s womb the best, and very young mice. Ulefnersen worked in older mice also, but not as well as it did in the younger mice. The first human that tried this drug was Jaci Hermstad, who had an identical twin sister who died of the same mutation as Jaci had. Jaci Hemstad ended up dying from ALS at age 26, but the biopsy revealed that mutant FUS protein was reduced significantly, which illustrates that the drug did what it was intended to do. She most likely died because she had already progressed too far into ALS when she received the drug, and it was shown that the drug worked best when younger mice received the drug over when older mice received the drug. She could have also died due to other factors such as mutant FUS was not the main contributor to the progression of the disease or environmental factors. This drug is now in phase 3 for clinical trials, a total of 77 patients will hopefully participate in the trial from all over the world, there are currently 18 patients participating globally.27
Therapies are also an option that can expand the life of a patient and slow the progression of the disease. They include speech therapy, occupational therapy, and physical therapy. Mental health care is also important in ALS care because ALS can cause depression, anxiety, and hopelessness, which could lead to faster progression in ALS. Breathing support is another tool that can expand the lifespan of a person with ALS even more than approved medications from the FDA. The different machines that can be used for breathing support are non-invasive ventilators, invasive ventilators, cough assisting machines, lung exercise machines, suction devices, and nebulizers. Ventilators decrease the amount of breathing you have to do on your own, by delivering positive-pressure ventilation. The difference between noninvasive ventilators and invasive ventilators is that non-invasive ventilators do not require a surgical incision and can be removed as needed or directed by a physician. Cough assist machines help patients cough out infectious agents since many ALS patients lose the ability to do so after a while. Lung exercise machines help improve lung function by expanding the lungs, so it is easier for patients to breathe. Suction devices are used to remove mucous and stuck food from the throat since ALS patients have limited ability to cough it up if it gets stuck in the throat. Nebulizers are used to deliver medications to ALS patients, if necessary if they already have asthma or acquire another infection where nebulizer medications would be beneficial.28 Even though there are very few options for some form of treatment for this specific mutation in ALS, it is hopeful that drugs will be approved to treat this illness.
Discussion:
In conclusion, c.1483 C>T, p.arg495 STOP FUS mutation in ALS is a very rare form of the disease and does not affect many people in the ALS population. This mutation is associated with familial ALS and juvenile ALS and is a very aggressive form of the illness. There is not an exact mechanism of how this mutation works in acquiring ALS, but there are many theories out there. Diagnosing this mutation for ALS comes with many challenges as well, such as the age range of patients, knowledge of neurologists and patients themselves, and timeline because this mutation is very rapid compared to others. There are not many options for treatment available currently, and none of the approved drugs from the FDA to slow the progression of ALS are proven to work for this mutation. Overall, ALS is a complicated disease and not easy to treat. This mutation is under-researched, which makes it harder to diagnose and treat compared to other mutations in familial ALS and sporadic ALS.
- ALS Facts and Symptoms Overview. (n.d.). ALS Pathways. Retrieved October 16, 2023, from https://www.alspathways.com/als-overview/; Amyotrophic Lateral Sclerosis: What Is It, Symptoms & Management. (n.d.). Cleveland Clinic. Retrieved October 19, 2023, from https://my.clevelandclinic.org/health/diseases/16729-amyotrophic-lateral-sclerosis-als ↵
- ALS Facts and Symptoms Overview. (n.d.). ALS Pathways. Retrieved October 16, 2023, from https://www.alspathways.com/als-overview/; Amyotrophic Lateral Sclerosis: What Is It, Symptoms & Management. (n.d.). Cleveland Clinic. Retrieved October 19, 2023, from https://my.clevelandclinic.org/health/diseases/16729-amyotrophic-lateral-sclerosis-als. ↵
- Is ALS Genetic? | The ALS Association. (n.d.). Retrieved October 20, 2023, from https://www.als.org/understanding-als/who-gets-als/familial ↵
- Marrone, L., Drexler, H. C. A., Wang, J., Tripathi, P., Distler, T., Heisterkamp, P., Anderson, E. N., Kour, S., Moraiti, A., Maharana, S., Bhatnagar, R., Belgard, T. G., Tripathy, V., Kalmbach, N., Hosseinzadeh, Z., Crippa, V., Abo-Rady, M., Wegner, F., Poletti, A., … Sterneckert, J. (2019) FUS pathology in ALS is linked to alterations in multiple ALS-associated proteins and rescued by drugs stimulating autophagy. Acta Neuropathologica, 138(1), 67–84. https://doi.org/10.1007/s00401-019-01998-x; Chen, C., Ding, X., Akram, N., Xue, S., & Luo, S.-Z. (2019). Fused in Sarcoma: Properties, Self-Assembly and Correlation with Neurodegenerative Diseases. Molecules, 24(8), 1622. https://doi.org/10.3390/molecules24081622 ↵
- ELLSSEE. (2015, July 29). Strength Tests if You Fear ALS for Legs & Upper Body. Scary Symptoms. https://scarysymptoms.com/2015/07/strength-tests-if-you-fear-als/ ↵
- Mackenzie, I. R. A., & Neumann, M. (2017). Fused in Sarcoma Neuropathology in Neurodegenerative Disease. Cold Spring Harbor Perspectives in Medicine, 7(12), a024299. https://doi.org/10.1101/cshperspect.a024299 ↵
- Mackenzie, I. R. A., & Neumann, M. (2017). Fused in Sarcoma Neuropathology in Neurodegenerative Disease. Cold Spring Harbor Perspectives in Medicine, 7(12), a024299. https://doi.org/10.1101/cshperspect.a024299; Bosco, D. A., Lemay, N., Ko, H. K., Zhou, H., Burke, C., Kwiatkowski, T. J., Jr, Sapp, P., McKenna-Yasek, D., Brown, R. H., Jr, & Hayward, L. J. (2010). Mutant FUS proteins that cause amyotrophic lateral sclerosis incorporate into stress granules. Human Molecular Genetics, 19(21), 4160–4175.https://doi.org/10.1093/hmg/ddq335.; Marrone, L., Drexler, H. C. A., Wang, J., Tripathi, P., Distler, T., Heisterkamp, P., Anderson, E. N., Kour, S., Moraiti, A., Maharana, S., Bhatnagar, R., Belgard, T. G., Tripathy, V., Kalmbach, N., Hosseinzadeh, Z., Crippa, V., Abo-Rady, M., Wegner, F., Poletti, A., … Sterneckert, J. (2019). FUS pathology in ALS is linked to alterations in multiple ALS-associated proteins and is rescued by drugs stimulating autophagy. Acta Neuropathologica, 138(1), 67–84. https://doi.org/10.1007/s00401-019-01998-x.; Ishigaki, S., & Sobue, G. (2018). Importance of Functional Loss of FUS in FTLD/ALS. Frontiers in Molecular Biosciences, 5, 44. https://doi.org/10.3389/fmolb.2018.00044. ↵
- What are Stress Granules? (2023, May 31). News-Medical.Net. https://www.azolifesciences.com/article/What-are-Stress-Granules.aspx.; Bosco, D. A., Lemay, N., Ko, H. K., Zhou, H., Burke, C., Kwiatkowski, T. J., Jr, Sapp, P., McKenna-Yasek, D., Brown, R. H., Jr, & Hayward, L. J. (2010) ↵
- Marrone, L., Drexler, H. C. A., Wang, J., Tripathi, P., Distler, T., Heisterkamp, P., Anderson, E. N., Kour, S., Moraiti, A., Maharana, S., Bhatnagar, R., Belgard, T. G., Tripathy, V., Kalmbach, N., Hosseinzadeh, Z., Crippa, V., Abo-Rady, M., Wegner, F., Poletti, A., … Sterneckert, J. (2019).; Liu, X., Chen, J., liu, W., Li, X., Chen, Q., Liu, T., Gao, S., & Deng, M. (2015). The fused in sarcoma protein forms cytoplasmic aggregates in motor neurons derived from integration-free induced pluripotent stem cells generated from a patient with familial amyotrophic lateral sclerosis carrying the FUS-P525L mutation. Neurogenetics, 16(3), 223–231. https://doi.org/10.1007/s10048-015-0448-y.; Chen, C., Ding, X., Akram, N., Xue, S., & Luo, S.-Z. (2019). Fused in Sarcoma: Properties, Self-Assembly and Correlation with Neurodegenerative Diseases. Molecules, 24(8), 1622. https://doi.org/10.3390/molecules24081622.; Ishigaki, S., & Sobue, G. (2018). Importance of Functional Loss of FUS in FTLD/ALS. Frontiers in Molecular Biosciences, 5, 44. https://doi.org/10.3389/fmolb.2018.00044.; Shellikeri, S., Karthikeyan, V., Martino, R., Black, S. E., Zinman, L., Keith, J., & Yunusova, Y. (2017). The Neuropathological Signature of Bulbar-onset ALS: A Systematic Review. Neuroscience and Biobehavioral Reviews, 75, 378–392. https://doi.org/10.1016/j.neubiorev.2017.01.045 ↵
- Alexander, E. J., Ghanbari Niaki, A., Zhang, T., Sarkar, J., Liu, Y., Nirujogi, R. S., Pandey, A., Myong, S., & Wang, J. (2018). Ubiquilin 2 modulates ALS/FTD-linked FUS-RNA complex dynamics and stress granule formation. Proceedings of the National Academy of Sciences of the United States of America, 115(49), E11485–E11494. https://doi.org/10.1073/pnas.1811997115.; Renaud, L., Picher-Martel, V., Codron, P., & Julien, J.-P. (2019). Key role of UBQLN2 in the pathogenesis of amyotrophic lateral sclerosis and frontotemporal dementia. Acta Neuropathologica Communications, 7(1), 103. https://doi.org/10.1186/s40478-019-0758-7 ↵
- Alexander, E. J., Ghanbari Niaki, A., Zhang, T., Sarkar, J., Liu, Y., Nirujogi, R. S., Pandey, A., Myong, S., & Wang, J. (2018). Ubiquilin 2 modulates ALS/FTD-linked FUS-RNA complex dynamics and stress granule formation. Proceedings of the National Academy of Sciences of the United States of America, 115(49), E11485–E11494. https://doi.org/10.1073/pnas.1811997115.; Renaud, L., Picher-Martel, V., Codron, P., & Julien, J.-P. (2019). Key role of UBQLN2 in the pathogenesis of amyotrophic lateral sclerosis and frontotemporal dementia. Acta Neuropathologica Communications, 7(1), 103. https://doi.org/10.1186/s40478-019-0758-7 ↵
- Sukhanova, M. V., Singatulina, A. S., Pastré, D., & Lavrik, O. I. (2020). Fused in Sarcoma (FUS) in DNA Repair: Tango with Poly(ADP-ribose) Polymerase 1 and Compartmentalisation of Damaged DNA. International Journal of Molecular Sciences, 21(19), Article 19. https://doi.org/10.3390/ijms21197020 ↵
- FYI: Criteria For the Diagnosis of ALS. (n.d.). The ALS Association. Retrieved October 20, 2023, from https://www.als.org/navigating-als/resources/fyi-criteria-diagnosis-als ↵
- Cedarbaum, J. M., Stambler, N., Malta, E., Fuller, C., Hilt, D., Thurmond, B., & Nakanishi, A. (1999). The ALSFRS-R: A revised ALS functional rating scale that incorporates assessments of respiratory function. Journal of the Neurological Sciences, 169(1), 13–21. https://doi.org/10.1016/S0022-510X(99)00210-5 ↵
- Obtaining an ALS Diagnosis. (n.d.). The ALS Association. Retrieved October 20, 2023, from https://www.als.org/navigating-als/resources/obtaining-als-diagnosis ↵
- Grad, L. I., Rouleau, G. A., Ravits, J., & Cashman, N. R. (2017). Clinical Spectrum of Amyotrophic Lateral Sclerosis (ALS). Cold Spring Harbor Perspectives in Medicine, 7(8), a024117. https://doi.org/10.1101/cshperspect.a024117.; Masrori, P., & Van Damme, P. (2020). Amyotrophic lateral sclerosis: A clinical review. European Journal of Neurology, 27(10), 1918–1929. https://doi.org/10.1111/ene.14393 ↵
- Grad, L. I., Rouleau, G. A., Ravits, J., & Cashman, N. R. (2017). Clinical Spectrum of Amyotrophic Lateral Sclerosis (ALS). Cold Spring Harbor Perspectives in Medicine, 7(8), a024117.https://doi.org/10.1101/cshperspect.a024117 ↵
- Masrori, P., & Van Damme, P. (2020). Amyotrophic lateral sclerosis: A clinical review. European Journal of Neurology, 27(10), 1918–1929. https://doi.org/10.1111/ene.14393 ↵
- Grad, L. I., Rouleau, G. A., Ravits, J., & Cashman, N. R. (2017). Clinical Spectrum of Amyotrophic Lateral Sclerosis (ALS). Cold Spring Harbor Perspectives in Medicine, 7(8), a024117. https://doi.org/10.1101/cshperspect.a024117 ↵
- Masrori, P., & Van Damme, P. (2020). Amyotrophic lateral sclerosis: A clinical review. European Journal of Neurology, 27(10), 1918–1929. https://doi.org/10.1111/ene.14393 ↵
- Grad, L. I., Rouleau, G. A., Ravits, J., & Cashman, N. R. (2017). Clinical Spectrum of Amyotrophic Lateral Sclerosis (ALS). Cold Spring Harbor Perspectives in Medicine, 7(8), a024117. https://doi.org/10.1101/cshperspect.a024117.; Masrori, P., & Van Damme, P. (2020). Amyotrophic lateral sclerosis: A clinical review. European Journal of Neurology, 27(10), 1918–1929. https://doi.org/10.1111/ene.14393 ↵
- Dormann, D., Rodde, R., Edbauer, D., Bentmann, E., Fischer, I., Hruscha, A., Than, M. E., Mackenzie, I. R. A., Capell, A., Schmid, B., Neumann, M., & Haass, C. (2010). ALS-associated fused in sarcoma (FUS) mutations disrupt Transportin-mediated nuclear import. The EMBO Journal, 29(16), 2841–2857. https://doi.org/10.1038/emboj.2010.143 ↵
- Neuralstem: Dr. Eva Feldman’s Latest Presentation on The Use of CUR’s Stem Cells in ALS (CUR, Buy, $4.04) | Expert Financial Analysis and Reporting | Smith on Stocks. (2014, April 11). https://smithonstocks.com/neuralstem-dr-eva-feldmans-latest-presentation-on-the-use-of-curs-stem-cells-in-als-cur-buy-4-04-paid-subscribers-only/ ↵
- Bellingham, M. C. (2011). A Review of the Neural Mechanisms of Action and Clinical Efficiency of Riluzole in Treating Amyotrophic Lateral Sclerosis: What have we Learned in the Last Decade? CNS Neuroscience & Therapeutics, 17(1), 4–31. https://doi.org/10.1111/j.1755-5949.2009.00116.x ↵
- MS, T. C. (n.d.). Radicava (edaravone) for ALS | ALS News Today. Retrieved October 20, 2023, from https://alsnewstoday.com/radicava-edaravone-for-als/ ↵
- MS, M. W. (n.d.). Relyvrio for ALS | ALS News Today. Retrieved October 19, 2023, from https://alsnewstoday.com/amx0035/ ↵
- Ionis Pharmaceuticals, Inc. (2023). A Phase 1-3 Study to Evaluate the Efficacy, Safety, Pharmacokinetics and Pharmacodynamics of Intrathecally Administered ION363 in Amyotrophic Lateral Sclerosis Patients With Fused in Sarcoma Mutations (FUS-ALS) (Clinical Trial Registration NCT04768972). clinicaltrials.gov. https://clinicaltrials.gov/study/NCT04768972.; Gagliardi, M., & Ashizawa, A. T. (2021). The Challenges and Strategies of Antisense Oligonucleotide Drug Delivery. Biomedicines, 9(4), 433. https://doi.org/10.3390/biomedicines9040433.; Ulefnersen. (n.d.). Ionis Pharmaceuticals, Inc. Retrieved October 19, 2023, from https://www.ionispharma.com/medicines/ion363/.; Promising ALS Therapy Moves Closer to Clinic. (2022, January 24). Columbia University Irving Medical Center. https://www.cuimc.columbia.edu/news/promising-als-therapy-moves-closer-clinic. ↵
- Managing Breathing Issues. (n.d.). The ALS Association. Retrieved October 19, 2023, from https://www.als.org/navigating-als/living-with-als/therapies-care/addressing-respiratory-changes/managing-breathing-issues.; Managing Breathing Issues | The ALS Association. (n.d.). Retrieved October 19, 2023, from https://www.als.org/navigating-als/living-with-als/therapies-care/addressing-respiratory-changes/managing-breathing-issues.; Braun, A. T., Caballero-Eraso, C., & Lechtzin, N. (2018). Amyotrophic Lateral Sclerosis and the Respiratory System. Clinics in Chest Medicine, 39(2), 391–400. https://doi.org/10.1016/j.ccm.2018.01.003 ↵


3 comments
michael
Very insightful article, hope more research can be done on ALS
Muskaan Tayal
This article was very interesting. I thought you did an amazing job!
Jake Gonzalez
Doing research of my own I’ve found some of the top basic knowledge you provided but I was never able to find an explanation specific to the FUS mutation. I appreciated the flow of the article and how nothing was too complicated for someone not as involved in biology.