Introduction
Stretchy skin does not sound like a primary symptom of a disease as much as it does a funny quirk of a character in a kids show, but in the case of Classical Ehlers-Danlos Syndrome (cEDS), stretchy skin is the hallmark of concern. Ehlers-Danlos Syndrome (EDS) is a family of connective tissue disorders that affects approximately one in every twenty-thousand people. Thirteen disorders are encapsulated in the family of EDS, some of which are more prevalent than others, but all are quite similar, differing in some of the presentations of different issues.1 cEDS is an intensely complicated and interwoven medical disorder that requires an equally intense examination to be truly understood. Breaking down cEDS requires the acknowledgement that this disease is a metaphorical Cerberus with three heads that each deserve their own examination. The genetic basis of cEDS is known but complex; various physical manifestations are truly germane to comprehend; and there is a breakdown of the management strategies that allow patients to live long and happy lives. To fully understand this uncommon but ever-present disorder, all three of these heads must be battled with and tamed to bring about a newfound knowledge of this disease.
Background
Because EDS affects the connective tissue of those afflicted, it is necessary to go over a slight amount of baseline information about what connective tissue is and what roles it plays in the body. At the most basic level, connective tissue is broken into two categories that are extremely key: connective tissue proper and specialized connective tissue. Of these two categories, the majority of EDS affects specialized connective tissue, which can further be broken down into a few categories that all need to be understood. First, adipose tissue is a type of specialized connective tissue that not only helps hold together different levels of the body, but this tissue is also the main location for fat storage, which acts as the body’s long-term energy storage for basic functions. Adipose tissue is subject to issues when EDS becomes involved, which can lead to different symptoms and presentations that will be discussed within this paper. The next type of specialized connective tissue is cartilage, which is one of the tissues most affected by EDS. Cartilage is what makes up the soft structures on the body, like the external ear and the nose. Cartilage also plays an immense role on the inside of the body, where it lines the joints and provides them with stability and cushion. The final two types of specialized connective tissue, bone and blood, are less affected by EDS, but because EDS is a disorder of connective tissue, there is still some effect on these categories.2 Proper connective tissue is also very important to understanding EDS. A large amount of proper connective tissue makes up our membranes and barriers between body parts, and a lot of proper connective tissue comes together to make tendons and ligaments that are particularly susceptible to EDS.3
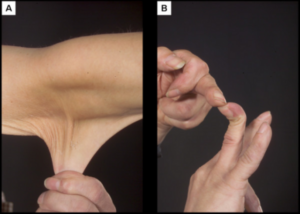
The other large piece of information needed to delve deeply into EDS is knowing how it is generally diagnosed. Taking the form of a three-step process, the diagnosis of EDS starts rather innocently with a full-body skin observation. Many things are looked for when examining the skin of a suspected patient. First, hyper-elastic skin is a very common marker for EDS. This is shown by either sagging skin in areas such as under the arms or behind the knee or by the skin on the body stretching out an inordinate amount. Next, atrophic scaring is noted anywhere it appears because the healing process involves the misfunctioning molecules of collagen in EDS patients. Finally, easy bruising is a trademark of these disorders that causes small patches of bruising, often on the hands or shins, with the patient having no recollection of the injury. If the physical examination discovers more than one of these telltale signs, they will move on to the second stage of testing, a skin biopsy.4
A skin biopsy is a small medical procedure where a small sample of skin is taken from the patient and visualized under a microscope. Using the microscope allows for intercellular structures to be seen and categorized. 5 The main structure to be on the lookout for with EDS is what is known as Collagen Flowers, which are malformed clusters of collagen fibers. Because EDS often affects collagen, it makes sense that this is the molecule of interest. The discovery of such structures warrants the procurement of a genetic test to confirm a mutation within one or more genes that are associated with the formation and behavior of collagen.6
Genetic Foundation
Approximately twenty genes are connected to the family of Ehlers-Danlos Syndrome. Every version of EDS has a different mixture of these genes mutated, but there is some commonality between all of them, and that is what proteins these genes encode for. Most of these genes affect the creation and processing of collagen types I, III, and V.7 These collagen types are all very prevalent in the human body, but type I is the most abundant protein in the entire human body. Making up 25-35% of all the fibrous proteins in the body, it is extremely crucial to the function of the human body.8
In the case of Classical Ehlers-Danlos Syndrome (cEDS), the main mutation comes on the COL1A1 gene, which codes for type I collagen. Specifically, the 134th amino acid of the alpha chain of type I collagen. This amino acid is transformed from an arginine to a cysteine, which is likely a single nucleotide switch from a cytosine or an adenine to an uracil. The recognition of this is incredibly important because this single nucleotide change causes an incredibly important protein to become malformed.9
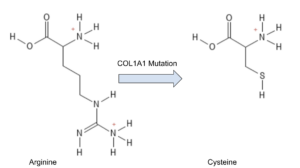
This change causes the arginine that has a positive charge at physiological pH to become neutral. Introducing a cysteine also presents a problem because cysteine residues can also link themselves together, forming disulfide bridges. This formation introduces new hyperstability to the protein that is in need of flexibility, but it also needs to be strong, so introducing unintended disulfide bonds and a loss of a positive charge is very dangerous.10 This seemingly small change seems to do a large amount of damage to the complex behaviors of the human body. Because this change takes place in one of the most produced proteins in the human body, the effect is seen in nearly every single bodily system.
Clinical Manifestations
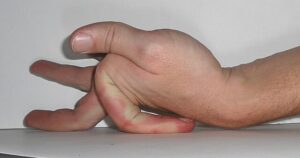
When presenting to a doctor with concerns of cEDS, or EDS in general, there are many things that the doctor should be looking for. First, the three most common symptoms that are involved are: hyper-elastic skin, when the skin stretches beyond what most would say is normal; atrophic scaring, which is a type of scar formation that involves indentation of the surrounding tissue; and hypermobile joints, the movement of joints being beyond a standard range of motion. These symptoms are almost always present in every person with EDS and warrant further investigation. There are some minor symptoms that are not always present but do provide further clinical support for an EDS diagnosis. These more minor symptoms include easy bruising, abnormal skin texture, and a history of joint dislocations.11
The presentation of EDS goes beyond the surface level and extends to the inside of the body as well. Digestive issues, muscular problems, and susceptibility to hernias are all common symptoms of various types of EDS. On the other hand, cEDS very rarely presents with internal symptoms until much later in life, when other bodily changes occur, such as old age causing degradation to the body.12 This is promising to those who are afflicted with cEDS because, although some of the surface-level issues can cause temporary issues and pain, there is a minimal threat of life-changing issues arising that could potentially end one’s life far too soon.
cEDS is, sadly, a lifelong disorder that will affect those afflicted as long as they live. There is no cure for this disease because it is the result of a genetic mutation, but because the symptoms experienced are generally mild, it often goes untreated, but the symptoms themselves can be attacked straight on.13
Treatment Strategies
Breaking down how to manage cEDS is, unfortunately, not about a cure but about the management of symptoms. Often, the skin issues are unable to be changed in any way that will make a large difference. Hyper-elasticity does not pose much of a threat to the everyday lives of those afflicted, so this is generally overlooked when it comes to management. Hypermobility of the joints is monitored to be sure that dislocations and other orthopedic issues do not arise. When it comes to the digestive issues associated with EDS, management can often entail a change in diet to adapt to low amounts of fermentable carbohydrates, which allows the body to not react as poorly with the excess gas created from the diet. The last major symptom, atrophic scaring, is only a cosmetic one, but if the scarring is widespread enough, it is possible for patients to seek out cosmetic surgery to mend it.14
There is a possibility that, in the future, there could be a way to avoid these types of diseases. Clustered Regularly Interspaced Short Palindromic Repeats (CRISPR) is an incredible new technology that is continually being researched. This technology allows biologists to edit the genome of cells while preserving their life sustainability accurately and precisely. This process is presently limited to smaller cell cultures, but the possibility of editing the genes of an embryo on one of its first cell divisions is becoming more feasible.15 EDS would be low on the priority list of genetic disabilities to prevent using CRISPR because of its low level of threat to life. Diseases like Huntington’s, Tay-Sachs, or sickle cell anemia that pose life-threatening issues will be a much higher priority. It is a truly exciting idea, but the prospect of editing the embryo of a human raises many ethical questions that would need to be fully examined before these types of experiments should be undertaken.
Conclusions
Classical Ehlers-Danlos Syndrome is a complex disease that affects many people worldwide, but knowing the foundational information about it can make the diagnosis less scary. There is potential for the eradication of EDS from the population in the coming years, but those who are afflicted with the disease will carry it for the rest of their lives. It is important for those affected to be diagnosed with the exact type of EDS that they are dueling with to gain the most accurate and successful management plan. More research is always needed on rare genetic disorders, and especially in this case, there are many pieces of the puzzle that are not yet understood. There is a vast amount of scientific brain power in the world, but only a very small amount of it is being used to investigate EDS. As far as chronic diseases go, EDS does not pose an incredible risk, but there is always concern for these diseases.
- Parapia, L. (2008, March 6). Ehlers-Danlos Syndrome—A Historical review. https://onlinelibrary.wiley.com/doi/full/10.1111/j.1365-2141.2008.06994.x ↵
- Kamrani, P., Marston, G., Arbor, T. C., & Jan, A. (2023). Anatomy, Connective Tissue. In StatPearls. StatPearls Publishing. http://www.ncbi.nlm.nih.gov/books/NBK538534/ ↵
- Fawcett, D. (2023, September 12). Connective tissue | Definition, Components, & Function | Britannica. https://www.britannica.com/science/connective-tissue ↵
- Sobey, G. (2015). Ehlers–Danlos syndrome: How to diagnose and when to perform genetic tests. Archives of Disease in Childhood, 100(1), 57–61. https://doi.org/10.1136/archdischild-2013-304822 ↵
- Ludmann, P. (2022, March 4). What is a skin biopsy? https://www.aad.org/public/diseases/a-z/what-is-skin-biopsy ↵
- Sobey, G. (2015). Ehlers–Danlos syndrome: How to diagnose and when to perform genetic tests. Archives of Disease in Childhood, 100(1), 57–61. https://doi.org/10.1136/archdischild-2013-304822 ↵
- Malfait, F., Castori, M., Francomano, C. A., Giunta, C., Kosho, T., & Byers, P. H. (2020). The Ehlers–Danlos syndromes. Nature Reviews Disease Primers, 6(1), Article 1. https://doi.org/10.1038/s41572-020-0194-9 ↵
- Stani, C., Vaccari, L., Mitri, E., & Birarda, G. (2020). FTIR investigation of the secondary structure of type I collagen: New insight into the amide III band. Spectrochimica Acta Part A: Molecular and Biomolecular Spectroscopy, 229, 118006. https://doi.org/10.1016/j.saa.2019.118006 ↵
- Nuytinck, L., Freund, M., Lagae, L., Pierard, G. E., Hermanns-Le, T., & De Paepe, A. (2000). Classical Ehlers-Danlos Syndrome Caused by a Mutation in Type I Collagen. American Journal of Human Genetics, 66(4), 1398–1402. ↵
- Rajpal, G., & Arvan, P. (2013). Chapter 236—Disulfide Bond Formation. In A. J. Kastin (Ed.), Handbook of Biologically Active Peptides (Second Edition) (pp. 1721–1729). Academic Press. https://doi.org/10.1016/B978-0-12-385095-9.00236-0 ↵
- Miklovic, T., & Sieg, V. C. (2023). Ehlers-Danlos Syndrome. In StatPearls. StatPearls Publishing. http://www.ncbi.nlm.nih.gov/books/NBK549814/ ↵
- Ritelli, M., Venturini, M., Cinquina, V., Chiarelli, N., & Colombi, M. (2020). Multisystemic manifestations in a cohort of 75 classical Ehlers-Danlos syndrome patients: Natural history and nosological perspectives. Orphanet Journal of Rare Diseases, 15(1), 197. https://doi.org/10.1186/s13023-020-01470-0 ↵
- Malfait, F., Castori, M., Francomano, C. A., Giunta, C., Kosho, T., & Byers, P. H. (2020). The Ehlers–Danlos syndromes. Nature Reviews Disease Primers, 6(1), Article 1. https://doi.org/10.1038/s41572-020-0194-9 ↵
- Brockway, L. (2016, January 4). Gastrointestinal problems in hypermobile Ehlers-Danlos syndrome and hypermobility spectrum disorders – The Ehlers-Danlos Support UK. https://www.ehlers-danlos.org/information/gastrointestinal-problems-in-hypermobile-ehlers-danlos-syndrome-and-hypermobility-spectrum-disorders/ ↵
- McCarty, N. S., Graham, A. E., Studená, L., & Ledesma-Amaro, R. (2020). Multiplexed CRISPR technologies for gene editing and transcriptional regulation. Nature Communications, 11(1), Article 1. https://doi.org/10.1038/s41467-020-15053-x ↵